
Summary
The last few decades have seen a massive rise in the amount of biomedical data, which has pushed the use of various Machine Learning (ML) approaches to solve new issues in clinical research and biological science. Artificial intelligence (AI) is revolutionizing bioinformatics by enabling the rapid analysis of complex and enormous biological data, the identification of hidden patterns, and the development of prediction models for numerous biological databases. ML and Deep Learning (DL) techniques make it possible to automatically extract features, choose which ones to utilize, and create predictive models, which makes it possible to research complicated biological systems effectively. This study intends to present an overview of DL so that bioinformaticians using these models can evaluate all relevant technical and ethical issues. The findings from this study will encourage people to use DL techniques to resolve their research questions while taking accountability, explainability, fairness, and potential biases. Finally, this study examines the changing environment of AI-driven tools and algorithms, emphasizing their critical role in accelerating research, improving data interpretation, and catalyzing discoveries in biomedical sciences.
Keywords
Artificial Intelligence (AI), Machine Learning (ML), Deep Learning (DL), and Natural language processing (NLP)
Artificial Intelligence in Bioinformatics
Background and Context
Bioinformatics is a multidisciplinary field that applies computational and analytical tools to acquire, process, and interpret biological data [1].This enables researchers to extract meaningful insights from complex biological information [1]. Rapid advancements in genomics, proteomics, and systems biology have led to an unprecedented influx of biological data generated through high-throughput sequencing, structural biology techniques, and computational methods. Among these advancements, breakthroughs in DNA sequencing technology have significantly enhanced the ability to decode genetic information, including both genomes and transcriptomes, in a cost-effective and time-efficient manner [2]. This progress has allowed scientists to investigate genetic landscapes beyond traditional model organisms, broadening the scope of biological research.
One of the primary applications of bioinformatics is genome analysis, which facilitates gene identification, functional annotation, and comparative genomic studies. Transcriptome sequencing plays a crucial role in genome editing by providing insights into gene expression, and genome sequence analysis enables precise genetic modifications [2]. Functional gene annotation helps identify key genetic elements and their roles, ultimately improving the accuracy and efficiency of genome-editing techniques [2]. Together, these tools advance personalized medicine, evolutionary studies, and drug discovery research.
Beyond its genomics and genome editing applications, it has become increasingly important in clinical decision-making and patient care. Clinical trials have indicated that pharmacist-led interventions, integrated with bioinformatics dashboards, have significantly reduced emergency department visits and hospitalizations while improving immune suppression monitoring [3,4]. These findings emphasize the value of bioinformatics-driven tools for optimizing healthcare strategies, enhancing treatment management, and improving patient outcomes. In addition to its clinical applications, bioinformatics is essential in proteomics, facilitating the analysis of protein expression, interactions, and functions in disease research and biomarker discovery. Advances in MS-based quantitative proteomics and computational predictions have identified functional peptides within proteins [5]. This approach has also supported in-silico proteolysis strategies, offering a promising method for enhancing the functional properties of protein hydrolysates [3][4]Bioinformatics also leverages systems biology and network-based approaches to unravel complex genes, proteins, and pathway interactions in chronic diseases, aiding the identification of key therapeutic targets. Additionally, it accelerates drug discovery by identifying promising treatment candidates, advancing personalized and effective medical interventions [4]. These wide-ranging applications underscore bioinformatics’ However, these datasets' sheer scale and complexity necessitate sophisticated computational approaches to ensure efficient processing, comprehensive analysis, and accurate interpretation.
Importance of Artificial Intelligence in Bioinformatics
Artificial Intelligence (AI) has revolutionized various fields, including bioinformatics, by bridging biological research with computational analysis. This integration enhances the ability to uncover hidden patterns and extract valuable insights from complex datasets more accurately and efficiently. Bioinformatics enables more effective data analysis, interpretation, and knowledge discovery by utilizing advanced AI-driven tools such as Machine Learning ML and Deep Learning (DL).AI surpasses traditional computational methods by employing adaptive algorithms that can dynamically process and analyze vast amounts of biological data. Unlike conventional approaches that rely on predefined rules and linear processing, AI continuously learns, identifies patterns, and refines its accuracy without explicit programming [4]. Automating data-driven decision-making and pattern recognition has significantly accelerated bioinformatics research, improving both speed and precision in scientific discoveries.
Scope and Objectives of the Review
This review comprehensively analyzes the evolving role and influence of AI in bioinformatics. It examines how various AI techniques, particularly ML, DL, and natural language processing (NLP), are leveraged to tackle complex biological challenges. Additionally, it highlights current trends, key methodologies, and emerging AI applications across multiple bioinformatics domains, including genomics, proteomics, and systems biology, focusing on areas such as genome analysis, protein structure prediction, and network modeling.
Beyond exploring applications, this review identifies challenges and limitations associated with AI in bioinformatics, such as data quality concerns, model interpretability, reproducibility issues, and integration with experimental approaches. It also delves into emerging trends and future research directions, including advancements in explainable AI, transfer learning, and novel applications in personalized medicine and synthetic biology.
Ultimately, this review aims to serve as a valuable resource for researchers, bioinformaticians, and AI practitioners, offering insights into how AI is reshaping bioinformatics and driving progress in biological research and healthcare.
Current Trends in AI for Bioinformatics
Machine Learning in Bioinformatics
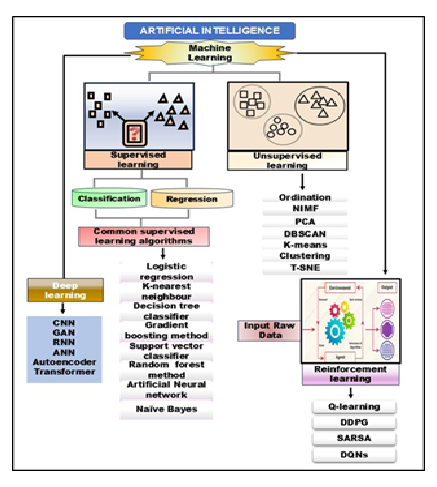
ML has transformed bioinformatics, which provides new tools and approaches to address intricate biological issues. Genomic sequences, protein structures, gene expressions, and clinical records are examples of the large, varied, and multifaceted data frequently found in bioinformatics. ML techniques have proven indispensable in analyzing and extracting significant patterns from these massive datasets. ML is used in bioinformatics for various purposes, from finding genetic variations linked to diseases to forecasting protein shapes and functions. Researchers can create more precise models for drug development, disease diagnostics, and biomarker discovery using supervised (for classification and regression task such as disease diagnosis and prognosis), unsupervised (for uncovering hidden patterns and patient subtypes), and reinforcement learning methods (for optimizing drug design and clinical trial strategies) is shown in Figure 1[5]. ML has played a pivotal role in advancing bioinformatics by developing sophisticated algorithms capable of handling large-scale biological data. Unlike conventional techniques such as molecular docking and sequence alignment—which are often labor-intensive and computationally expensive—ML-based approaches offer more efficient and scalable solutions. However, more precise classifications and predictions are made possible by the ability to train ML models to recognise patterns in data. DL, a type of machine learning, has shown remarkable results in proteomics and genomics. Deep neural networks (DNN), for example, had previously been assumed to be incapable of accurately predicting proteins' secondary and tertiary structures. Similarly, ML algorithms may examine transcriptome data to provide information about the control of gene expression and how it relates to different diseases, thereby supporting the developing field of personalised medicine. Additionally, ML is becoming increasingly important in developing and discovering new drugs. ML approaches speed up the possible identification of medication candidates, whereas the traditional drug development procedure is expensive and time-consuming. ML models can anticipate a compound's biological activity by analysing chemical databases, simplifying the early drug creation phases. Furthermore, ML is essential to precision medicine because it makes it possible to create algorithms that, using a patient's genetic composition, can forecast how they will react to treatment. Combining these technologies makes it feasible to adopt more individualised therapy strategies, increasing treatment effectiveness while reducing adverse effects [6]. ML encompasses a broad range of techniques that enable computers to learn patterns from data and make informed decisions or predictions. In bioinformatics, ML is increasingly used to analyze complex and high-dimensional biological data. Depending on the nature of the data and the specific task, following learning paradigms can be applied:
-
Supervised Learning
These algorithms use labeled training data to learn a function that maps input data to desired output labels. Examples include decision trees, support vector machines (SVMs), and linear regression [6]. -
Unsupervised Learning
These algorithms do not use labeled training data and instead try to identify patterns and relationships in the data. Examples include clustering algorithms (e.g., k-means), dimensionality reduction algorithms (e.g., principal component analysis), and anomaly detection algorithms [6]. -
Reinforcement Learning
These algorithms involve an agent learning to interact with its environment to maximize reward. These algorithms are used in bioinformatics for protein folding and drug design tasks, as described in Figure 1 [5]. -
Semi-supervised Learning
This involves training a ML model on a partially labeled dataset to use the labeled examples to make predictions about the unlabeled samples [5].
Deep Learning (DL) in Bioinformatics
DL has emerged as a transformative approach in the field of bioinformatics, offering powerful tools to analyze and interpret complex biological data [1]. By learning from vast amounts of genomic, proteomic, and clinical data, ML algorithms can assist in tasks such as disease classification, biomarker identification, drug discovery, and personalized medicine. Its ability to adapt and improve from new data makes ML particularly valuable in handling the dynamic and high-
Figure 1: Overview of Artificial Intelligence and ML Paradigms: This figure illustrates the hierarchical relationship between AI, ML, and its three primary branches—Supervised Learning, Unsupervised Learning, and Reinforcement Learning. It highlights key algorithms and techniques within each branch, including DL models (CNN, GAN, RNN, ANN, Autoencoder, Transformer), standard supervised learning algorithms (e.g., logistic regression, decision trees, SVM, neural networks), unsupervised learning methods (e.g., PCA, K-means, clustering), and reinforcement learning strategies (e.g., Q-learning, DDPG, SARSA, DQNs).
dimensional nature of biological datasets. Traditional ML techniques often require manual feature engineering, which can be time-consuming and challenging. In contrast, DL models are capable of automatically learning high-level features directly from raw data, reducing the need for extensive human intervention. [7]. When a problem can be effectively addressed through a well-defined mathematical model, the application of ML is typically redundant. Nevertheless, biology involves a complicated interaction of multiple factors that mathematical formulas cannot entirely express. Therefore, it makes sense to use machine learning, especially DL. Traditional ML techniques, particularly in gene expression research, often rely on manual feature curation—requiring domain experts to identify, select, and engineer relevant features from raw data. This process can be both time-consuming and prone to bias, as it depends heavily on prior biological knowledge and assumptions about which features are most informative for a given predictive task [8]. This is somewhat simple for gene expression, but it is more difficult to determine if an RNA sequence is a pre-microRNA. It is necessary to manually select thousands of attributes and determine whether each is pertinent [9]. Under such circumstances, DL is particularly relevant to bioinformatics since it can directly learn higher-level features from the data [10]. These algorithms leverage DNN to learn complex patterns and relationships within data. Alternatively, for a more formal or academic tone: By employing DNN, these algorithms are capable of capturing intricate patterns and dependencies in biological data. This capability has enabled significant advancements in various bioinformatics applications such as drug discovery, protein structure prediction, and the analysis of gene expression profiles
-
Deep Neural Networks
At the core of DL’s success in bioinformatics are DNNs, which enable the modeling of complex biological processes through multiple layers of interconnected neurons [11]. Several studies have employed more straightforward methods, such as forecasting protein secondary structures or torsion angles; however, fully predicting protein conformations in three-dimensional space remains a challenging and complex task. For instance, stacked autoencoders (SAEs) have been used to address prediction problems related to accessible surface area, torsion angles, and secondary structures within protein amino acid sequences [12]. In a different investigation, Spencer et al. used Deep Belief Networks (DBN) in conjunction with Position Specific Scoring Matrix (PSSM) and Atchley factors to predict protein secondary structure [11].
DNNs have proven highly effective in deciphering the intricate mechanisms underlying gene expression regulation. For example, Lee et al. proposed a novel training method for DBNs called boosted contrastive divergence, specifically designed to handle imbalanced data, along with a new regularization term to promote sparsity in DNA sequence representations. They applied this approach to splice junction prediction and demonstrated significantly enhanced performance [7].
Furthermore, Chen et al. used a multi-layer perceptron (MLP) to estimate the expression of up to 21,000 target genes from just 1000 landmark genes using microarray and RNA-seq expression data [13]. The skip-gram model, a variation of MLP, was used to classify proteins and learn distributed representations of biological sequences. Fakoor et al. utilized SAEs to classify various types of cancer and applied PCA for dimensionality reduction in anomaly detection tasks [14]. -
Convolutional Neural Networks (CNNs)
Although only a limited number of studies have employed CNNs to address biological sequence problems—particularly in gene expression regulation—these works have highlighted CNNs’ strong potential. One key advantage is that PSSMs are learned directly from data. Initial convolutional layers act as motif detectors capturing local sequence patterns, and deeper layers learn more complex features.
CNNs are also suitable for multitask cooperative learning. One example transformed ChIP-seq data into a 2D matrix and applied a CNN where each row represented transcription factor activity. More recent research has focused on 1D CNNs directly applied to sequence data. CNN-based methods have been proposed for transcription factor binding site prediction and cell-specific DNA accessibility multitask prediction [15].
Zeng et al. (2016) [16] showed the importance of the number of filters over layers in motif-based tasks. Zhou et al. (2015) introduced DeepSEA, a CNN-based framework for predicting expression quantitative trait loci (eQTLs) and disease-associated variants via multitask joint learning across chromatin features [17]. -
Recurrent Neural Networks (RNNs)
Given the sequential nature of biological data, RNNs are ideal for tasks such as protein classification and gene expression regulation. Early studies used Bidirectional RNNs (BRNNs) with perceptron units to predict protein structure. Sønderby et al. later used BRNNs with LSTM units and 1D convolutions to classify protein subcellular localization [18].
Lee et al. used LSTM-based RNNs for microRNA identification and target prediction, significantly improving performance over prior methods [19]. -
Emergent Architectures
Recent protein structure prediction advances have adopted novel architectures. In 2017, Min et al. [20] used Deep Spatio-Temporal Neural Networks (DST-NNs), leveraging spatial features like alignment and orientation probabilities. Multi-Dimensional RNNs (MD-RNNs) were also employed to model dependencies in secondary structures, correlation profiles, and amino acid sequences, improving predictive performance.
| DL | Omics | Biomedical Imaging | Biomedical Signal Processing |
|---|---|---|---|
| Deep neural networks |
Protein structure, Gene expression regulation, Protein classification, Anomaly classification |
Anomaly classification, Segmentation, Recognition, Brain decoding |
Brain decoding, Anomaly classification |
| Convolutional neural networks | Gene expression regulation |
Anomaly classification, Segmentation, Recognition |
Brain decoding, Anomaly classification |
| Recurrent neural networks |
Protein structure, Gene expression regulation, Protein classification |
Brain decoding, Anomaly classification |
|
| Emergent architectures | Protein structure | Segmentation | Brain decoding |
Table 1: DL applied bioinformatics research avenues and input data
Natural Language Processing (NLP) in Bioinformatics
Natural Language Processing (NLP) is an interdisciplinary field that bridges artificial intelligence and linguistics, focusing on the development of computational tools capable of interpreting, processing, and generating large volumes of human language data. The complexity of natural language analysis arises from its nuanced semantics, where word order and contextual meaning both play critical roles—a single sentence can convey multiple interpretations depending on the surrounding context. Although biological sequences lack explicit semantic structures like those in human languages, this review demonstrates that NLP techniques can still yield meaningful insights when applied to biomolecular data. Our primary objective is to explore the application of NLP algorithms in bioinformatics, beginning with text mining in PubMed abstracts and extending to the analysis of nucleic acid and protein sequences. The approaches discussed are primarily based on word2vec [21] and transformer-based architectures [22], which have shown promise in capturing patterns and relationships in biological data.
-
(a)Word2Vec
While natural language text cannot be directly input into neural networks without mathematical preprocessing, Word2Vec was developed based on this foundational concept. One of the most widely used approaches for converting textual data into numerical form is the creation of n-dimensional vectors—commonly referred to as word embeddings. This technique effectively addresses the challenge of capturing semantic relationships between words.
A well-known example illustrating this principle involves the words king, queen, man, and woman. Ideally, in the embedding space, the relationship between king and queen mirrors that between man and woman. Mikolov et al. demonstrated this with the following vector arithmetic formula:vector(“King”) − vector(“Man”) + vector(“Woman”) = vector(“Queen”) [21]
"Embedding" refers to the process of converting data—such as words—into vector representations in a continuous, high-dimensional space. The Word2Vec model is composed of three main components:- An input layer
- An output layer
- A hidden layer (embedding layer)
- Continuous Bag-of-Words (CBOW): Predicts a target word based on its surrounding context.
- Skip-Gram: Predicts the surrounding context words based on a given target word; essentially the inverse of CBOW.
-
(b)Transformers
In 2017, Vaswani et al. introduced the Transformer architecture as a breakthrough solution to several limitations faced by traditional models like RNNs in processing natural language texts [23].
Transformers overcame key challenges such as:- Limited parallelization during training
- Difficulties in capturing long-range dependencies due to memory constraints
- Fixed input sequence lengths
The standard Transformer architecture is composed of two primary components:- Encoder: Processes the input sequence
- Decoder: Generates the output sequence [24]
Input: The initial step involves vectorizing the input data—typically textual data—to generate embeddings that represent words or tokens in a continuous vector space.
Positional Encoding: Since Transformers do not rely on recurrence or convolution to capture sequence order, positional encoding is introduced to inject information about the position of tokens within a sequence. These positional encodings are typically created using sinsusoidal functions of different frequencies, producing unique vectors for each position. The positional encoding vectors are then added element-wise to the input embedding, combining with positional information. This enables the model to distinguish token order and capture the sequential nature of the data, which is crucial for understanding context
Encoder: The encoder processes the sum of the input embeddings and their positional encodings. It comprises two key sub-layers:
Multi-Head Attention (MHA): This mechanism allows the model to simultaneously attend to different positions in the sequence to capture contextual relationships.
Feed-Forward Neural Network (FFN): A fully connected layer that applies nonlinear transformations to the output of the MHA Residual (bypass) connections are incorporated between layers to preserve original input information and improve gradient flow. Each sub-layer is followed by layer normalization to stabilize and speed up training.
Masked Multi-Head Attention: Ensures that predictions for a particular position only consider known outputs up to that point, maintaining autoregressive properties.
Encoder-Decoder Attention (MHA): Allows the decoder to focus on relevant parts of the encoder’s output.
Feed-Forward Network and Residual Connections: Similar to the encoder, the decoder uses FFNs and residual connections, followed by layer normalization.
Final Prediction: The decoder's output undergoes a linear transformation before being passed through a softmax layer. The softmax function produces a probability distribution over the output vocabulary, with each value indicating the model’s confidence in predicting the corresponding token [25].
Restructuring of Core ML Techniques – Applications in Bioinformatics SubfieldsOn establishing foundational ML methodologies, their transformative role in key bioinformatics domains is now explored. Our interpretation of biological data, from genomics to structural biology, is now being reshaped by AI-driven approaches, thus enabling insights at unprecedented scale and precision as depicted in Figure 2.
Role Of AI In Genomics and Epigenomics
Genomics is a multidisciplinary field dedicated to understanding the structure, function, and evolution of genomes by applying advanced sequencing technologies and bioinformatics tools [30]. By exploring the entirety of an organism's genetic material, genomics seeks to uncover the intricate relationships between genome organization, gene function, and evolutionary processes [31]. The field is broadly divided into several specialized areas, including structural genomics, which focuses on the organization and physical structure of the genome, and functional genomics, which investigates the roles and regulatory mechanisms of genes and non-coding regions.
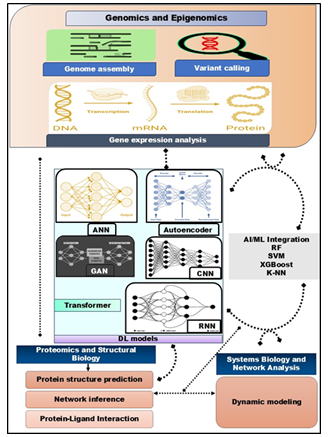
Figure 2. Integrated Multi-Omics and DL Framework: Comprehensive framework illustrating the integration of genomics, epigenomics, and gene expression analysis with advanced DL and artificial in (AI/ML) models-including ANN, GAN, CNN, RNN, autoencoders, and transformers—for downstream applications in proteomics, structural biology, and systems biology. The diagram highlights how these computational approaches enable protein structure prediction, network inference, protein-ligand interaction studies, and dynamic modeling for holistic biological and biomedical research.
Advances in next-generation sequencing (NGS) are used by researchers to sequence DNA/RNA with high accuracy and detect genetic variants/mutations [32]. Eepigenomics is the study of mechanisms associated with changes in gene expression without changing the DNA sequence, influenced by factors such as environmental conditions, lifestyle, and disease state. In this process, a complex interaction occurs between genotypes of an individual and the surrounding environment, which plays a pivotal role in disease development [29]. The modifications involved in this are DNA methylation, histone modifications, and small non-coding RNAs, which are major factors responsible for the activation or repression of genes [34]. Epigenetic biomarkers—particularly DNA methylation patterns—hold significant promise in clinical research and practice due to their potential roles in early disease detection, diagnostic precision, and therapeutic monitoring. Their stability and detectability in biological samples, even at early stages of disease, make them especially valuable in clinical applications. Recent advancements in single-cell epigenomics, combined with AI-driven omics technologies, are further accelerating the integration of genomic and epigenomic markers into personalized medicine.
Genome assembly
Genome assembly is a fundamental process in genomics that involves aligning and merging short DNA sequence reads to reconstruct the original, continuous genomic sequence of an organism. It serves as a critical foundation for downstream genomic analyses, enabling the study of gene structure, function, and evolutionary relationships [35] [36]. Next-Generation Sequencing (NGS) techniques have revolutionized genomic research through their high-throughput capability, allowing millions of sequencing reactions to occur simultaneously. The present NGS platforms used are Illumina [37], Ion Torrent, and sequencing by Oligonucleotide Ligation and Detection (SOLiD)[38]. These technologies offer distinct strategies, with specific advantages and disadvantages. Most of the NGS platforms generate short read lengths of less than 300 base pairs, which complicates de novo genome assembly, making resolving repetitive regions and achieving contiguous sequences difficult [39]. These platforms also face challenges in accurately sequencing regions with extremely high G+C content, as well as tandem and interspersed repeat sequences. These sequencing platforms often encounter difficulties in accurately reading regions with extremely high G+C content, as well as tandem and interspersed repeat sequences, which can result in such regions being underrepresented or entirely missing from sequencing datasets. Fragmentation and incomplete assemblies are additional common challenges associated with these technologies. To address these limitations, genome assembly strategies increasingly employ hybrid approaches that combine short-read NGS data with long-read sequencing technologies, such as PacBio or Oxford Nanopore. These integrated methods enhance assembly accuracy and completeness by leveraging the strengths of each platform. As NGS technologies continue to advance, the primary challenge has shifted from data generation to the efficient analysis, integration, and assembly of vast genomic datasets. [40] A variety of AI-powered tools are being leveraged to enhance genome assembly and analysis. For instance, Seq2Squiggle utilizes a Feed-Forward Transformer (FFT) architecture to simulate nanopore sequencing signals directly from nucleotide sequences[41]. Unlike autoregressive models, this approach processes inputs in parallel, resulting in faster and more stable signal prediction. It employs multi-head attention and dense layers to effectively extract sequence features for precise signal mapping. A length regulator dynamically expands DNA embeddings to align with the expected duration of nanopore signals, using a gamma distribution model to adjust event lengths. Additionally, a noise sampler introduces Gaussian noise to mimic the natural variability observed in real sequencing data. By addressing the limitations of traditional simulators—such as k-mer–based methods that struggle to adapt to evolving nanopore chemistries—this tool offers improved flexibility and accuracy. It shows strong potential for optimizing benchmarks on Nanopore R10.4 and R9.4.1 flow cell technologies, making it highly relevant to current research needs. [41]. It uses a Feed Forward Transformer (FFT) to generate realistic nanopore sequencing signals from DNA sequences. Another DL-based diploid consensus tool, CONNET, has been developed to enhance both the efficiency and accuracy of genome assembly from long-read sequencing data. This tool addresses the high error rates commonly associated with long reads by leveraging spatial relationships within the alignment pile-up to improve consensus accuracy. A sliding window of size three is employed for improved tensor feature extraction. CONNET utilizes a BRNN with one fewer layer than Medaka yet achieves superior accuracy. The input tensor effectively captures alignment features, enabling more efficient neural network learning. CONNET was evaluated on multiple datasets, including E. coli SCS110 (90 datasets) sequenced with R9.4.1 chemistry, E. coli K-12 (174 datasets) with R9 chemistry, and Homo sapiens (37 datasets) also with R9.4.1 chemistry, producing high-quality diploid genome consensus results.
Another notable tool, SPAdes (St. Petersburg genome Assembler), was originally developed for de novo assembly of genome sequencing data from cultivated microbial isolates and single-cell genomics DNA sequencing [42]. It was enhanced through hybrid assembly approaches that combine short reads (IonTorrent) with long reads (Oxford Nanopore). This tool supports five distinct pipelines tailored for genome assembly, metagenomics, transcriptomics, plasmid reconstruction, and biosynthetic gene cluster analysis from both metagenomic and whole-genome datasets. AI-driven modifications to SPAdes have further improved metagenomic assembly and classification by boosting computational efficiency and increasing assembly accuracy [42].
Variant calling
Variant calling is the process of detecting genetic variations, including single-nucleotide variants (SNVs) and short insertions or deletions (indels), from sequencing data. Over the years, this field has seen substantial advancements, with next-generation sequencing (NGS) technologies and advanced computational algorithms greatly enhancing both the accuracy and efficiency of variant detection[43]. Genetic variations refer to differences in DNA sequences that arise within a species or between different species. Next-generation sequencing (NGS) platforms, such as Illumina, represent second-generation technologies, while third-generation platforms include Pacific Biosciences and Oxford Nanopore Technologies [44,32]. Third-generation sequencing makes sample preparation easier and yields longer reads—often several kilobases—by enabling real-time sequencing of individual DNA molecules without amplification [45]. Oxford Nanopore’s MinION exemplifies nanopore sequencing technology, which detects nucleotide sequences by monitoring changes in ionic current as DNA molecules pass through nanopores. The use of hairpin adapters enables sequencing of complementary strands, thereby enhancing both accuracy and efficiency [43].
Computational methods like the MinKNOW platform provide high-accuracy sequencing reads and have been successfully applied in pathogen identification, such as detecting the Ross River virus with over 98% accuracy within hours. The combination of accuracy and portability holds great promise for advancing both research and clinical diagnostics.
DeepVariant, developed by Google AI, is a state-of-the-art DL tool designed for highly accurate variant calling from NGS data. Unlike traditional rule-based bioinformatics tools, DeepVariant employs a deep convolutional neural network (CNN) to transform raw sequencing data into high-confidence genetic variant calls. This learning-based approach enables it to perform consistently across various sequencing platforms—including Illumina, PacBio, and Oxford Nanopore—accurately identifying single-nucleotide variants (SNVs) and insertions/deletions (Indels). Its platform-agnostic design makes it highly versatile for applications in population genetics, cancer genomics, and rare disease research.
An extension of DeepVariant, called DeepTrio, incorporates parental data to improve detection of de novo mutations in family-based sequencing, further showcasing the tool’s adaptability. Additionally, DeepVariant demonstrates robust performance even with low-coverage or noisy data, making it a powerful asset for high-throughput genomic studies.
In the context of ovarian failure, AI-driven blood-based gene variant profiling utilized Whole Exome Sequencing (WES) combined with MLmodels like Random Forest and unsupervised clustering. Analyzing 63,928 genetic variants, this approach identified 116 variants with significant allele frequency differences and classified ovarian failure into two genomic subtypes (A & B) with 97.2% accuracy. Similarly, bioinformatics and ML-based studies on Premature Ovarian Failure (POF) employed WES alongside tools such as VEST and CADD, uncovering nine heterozygous variants in 24% of patients. Key genes linked to DNA repair and infertility—including MCM8, MCM9, EIF2B3, PREPL, ERCC6, and HFM1—were identified, along with 72 novel variants potentially involved in folliculogenesis
[47]. Beyond reproductive health, AI has been leveraged to predict hypertension risk by applying ML models trained on genetic variants and gene expression data. Analysis of Whole Genome Sequencing (WGS) data using SVM and LR showed that Linear SVM achieved the highest predictive performance, with an AUC of 0.777. Interestingly, incorporating gene expression data reduced the predictive accuracy, suggesting that genetic variants alone may provide a more reliable basis for hypertension risk assessment.
AI also plays a vital role in predicting bacterial pathogenicity. For example, in a study on Listeria monocytogenes, supervised ML models—including SVM, Random Forest (RF), Neural Networks, and Gradient Boosting—were used to analyze virulence genes. The linear SVM model reached an accuracy of 89% in identifying virulent strains. Key genes such as InlK, InlJ, InlF, FAM002725, and lmo2026 were associated with foodborne outbreaks and the severity of disease [48].
Overall, while AI does not directly perform raw variant calling, it significantly enhances variant analysis by using ML models to interpret and classify genetic variants obtained from Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS). These models play a crucial role in disease classification, biomarker discovery, and risk prediction. Collectively, these applications highlight AI’s transformative impact on genomics, improving the accuracy, efficiency, and scalability of variant analysis and disease diagnostics, thereby advancing the field of precision medicine. By measuring the transcriptional output of genes, gene expression analysis is an essential next step that connects genetic diversity to subsequent molecular and cellular effect. By combining gene expression profiling and variant calling, researchers can uncover regulatory mechanisms, link genotype to phenotype, and find biomarkers associated with both health and disease. The next section examines the ways in which artificial intelligence has improved gene expression analysis, allowing for deeper comprehension of gene regulation in intricate biological contexts and more precise interpretation of transcriptomic data.
Gene expression analysis
Gene expression analysis quantifies the activity of thousands of genes simultaneously, providing insights into cellular and molecular functions and imparting information about disease mechanisms.[49]. AI-based approaches have significantly enhanced expression analysis by enhancing the accuracy of data interpretation, reducing technical variability, and identifying novel biomarkers. AI algorithms play a crucial role in profiling gene expression datasets extracted through high-throughput sequencing techniques, such as microarrays and RNA sequencing. These methods enhance classification, pattern recognition, and feature selection, enabling researchers to differentiate between different biological states, diseased and control conditions. ML techniques, such as supervised and unsupervised learning, are widely used in gene expression analysis studies. For the classification of gene expression profiles, ML algorithms are employed, including Gradient Boosting and Extreme Gradient Boosting (XGBoost), RF, and k-NN [50,51,52,53,54,55].Dimensionality reduction techniques such as PCA and t-SNE help in visualization and manage high-dimensional gene expression data[56].
Kernel-based methods, such as SVM, efficiently classify DEGs and help differentiate healthy and diseased datasets. Gaussian Process Classification (GPC) further enables the modelling of non-linear relationships and uncertainties in gene expression datasets [57]. DL models such as autoencoders and CNNs extract meaningful features from complex datasets in transcriptomics profiling. Transformer-based architectures, inspired by NLP, have also been adapted for analyzing single-cell RNA sequencing (scRNA-seq) data, improving rare cell-type identification and gene regulatory network reconstruction[58].The following case studies illustrate the diverse and impactful applications of AI-driven approaches in gene expression analysis.
Case Studies on AI in gene expression analysis
Cancer Subtype Identification Using Bayesian Neural Networks
For efficient, individualized treatment, it is essential to accurately identify the subtypes of cancer. Uncertainty is a problem for traditional classifiers, particularly when subtypes are quite close.
EpICC, a Bayesian neural network-based classifier created by Joshi et al., measures epistemic uncertainty in its predictions in addition to predict cancer subtypes. Because the model includes an uncertainty correction step, situations that are unclear can be marked for additional verification rather than being rejected. In terms of overall classification accuracy, EpICC fared better than current techniques. But it has trouble discriminating apart very similar subtypes. It was proposed that including multi-omics data, including epigenetic alterations, would increase precision even more. The model's capacity to measure uncertainty is especially useful for applications using liquid biopsies and other non-invasive cancer detection techniques where accurate categorization is crucial [59].
Cancer Grade Prediction Using Gradient Boosting Trees
One important predictive marker for breast cancer is its histological grading, however manual grading can be arbitrary and unreliable.Amiri Souri et al introduced the Cancer Grade Model (CGM) based on GBT trained on microarray data from 5,031 untreated breast cancers spanning 33 published datasets, and corresponding clinical data were integrated. The model was trained on histological grade-1 and grade-3 samples and then applied to grade-2 and unknown-grade samples for prognostic risk classification[60]. CGM showed strong efficacy in prognostic risk stratification and cancer grade classification, facilitating more objective and repeatable grading. This strategy can help pathologists provide reliable prognostic evaluations, ultimately enhance patient management.
ML-Based Classification of Neurodegenerative Diseases (NDDs)
It can be difficult can be difficult to diagnose NDDs like Parkinson’s disease (PD) and Alzheimer’s disease (AD) early and accurately since their clinical symptoms overlap. Using blood-based biomarkers data from 377 individuals, Lin CH et al used ML models to measure Aβ42, Aβ40, total tau, p-Tau181, and α-synuclein. Linear Discriminant Analysis (LDA) model was used to extract feature, and several classifiers were evaluated. RF had the best accuracy up to 76% in differentiating between NDD’s, while AD had an accuracy of 83%
[61].
Gene Expression-Based Lung Cancer (LC) analysis
Globally, LC continues to be major cause of cancer-related mortality. Finding therapeutics targets and comprehending tumor progression depends on identifying DEGs. High-dimensional RNA-seq data frequently presents challenges for conventional statistical techniques. This study analyzed RNA-seq data from LC samples (NCBI SRP009408) using multi algorithms framework. RF, Lasso, XGBoost, Gradient Boosting Elastic Net, and MLP, SVM, and k-NN were applied to identify robust DEGs. The ensemble approach prioritized genes consistently flagged across models to reduce false positives. This study highlights the top five up-regulated genes COL11A1, TOP2A, SULF1, DIO2, MIR196A2) and top five downregulated genes are PDK4, FOSB, FLYWCH1, CYB5D2, MIR328[62].
Sepsis Classification Using ML Models
Conventional methods that rely on nonspecific biomarkers and clinical ratings frequently impede the timely diagnosis of sepsis, a life-threatening illness that necessitates prompt treatment. Using gene expression data from sepsis patients and controls from the GEO and EMBL-EBI Array databases, a study classified sepsis and identified DEGs using DT, RF, SVM, and DNN. With an accuracy of 89%, the models outperformed conventional statistical methods (72%), identifying 2,361 significant DEGs, including important genes like S100A8 (related with inflammatory response) and CD177 (associated with neutrophil activation). The identified DEGs may help guide fast diagnostic panels or treatment targets, and our ML-driven method allows for earlier, more accurate sepsis diagnosis, potentially lowering mortality rates [63].
While gene expression analysis provides us knowledge about the cellular activity, understanding of protein dynamics is equally important for converting data into functional biology as gene expression analysis. This leads us to role of AI in proteomics and structural biology.
Role of AI in proteomics and structural biology
Proteomics and structural biology are important for understanding the functional and structural dynamics of protein, which are the centre of virtually all biological processes. Proteomics focuses on the large-scale study of proteins, covering their expression, post translational modifications, interactions, and functions[64].Recent advances in MS and high-throughput sequencing technologies have enabled comprehensive proteomic profiling, facilitating biomarker discovery, disease characterization, and drug target identification. Structural biology, in parallel, aims to elucidate the three-dimensional (3D) architecture of biomolecules, primarily utilizing techniques such as cryo-EM [65], [66], [67]. Understanding protein structure at atomic resolution is essential for deciphering molecular mechanisms, protein-ligand interactions, and for rational drug design.
Despite remarkable experimental progress, many challenges persist, including the high cost, time consuming nature, and technical limitations of traditional structure determination methods. In this context AI has emerged as a transforming force, enabling unprecedented insights into protein function, interaction networks, and structure-based drug discovery. The following sections explore how AI is revolutionizing both proteomics and structural biology.
Protein Structure Prediction (PSP)
Predicting protein structures from amino acid sequences has been a persistent challenge in bioinformatics and biochemistry. Accurate structure prediction is important for understanding protein function, guiding drug designing, and developing novel therapeutics. Traditional computational approaches such as homology modeling, molecular dynamics, are often limited by high computational costs, time requirements, and restricted accuracy, especially for proteins lacking homologous templates. Recent advances in AI, particularly DL, have dramatically improved the accuracy and speed of PSP. Early DL models, such as CNNs were used to predict contact maps indicating spatial proximity between amino acid residues. These models input features, and predicts spatial relationship, which are then converted into full 3D atomic structures using gradient-based optimization. Graph Neural Networks (GNNs) and RNNs further enhanced the ability to capture long-range dependencies in protein sequences and structure [68] .Attention mechanisms, especially in transformer architecture, have enabled the more effective use multiple sequence alignments (MSAs) and structural templates.
Among the most promising AI-based tools is Alphafold2, developed by DeepMind[69]. Alphafold 2 utilizes transformer neural network, and graph-based reasoning on structural data from the Protein Data Bank (PDB) to predict 3-D conformations of proteins [70]. Although Alphafold 2 has attained near experimental accuracy for many single chain proteins, it still has issues with dynamic conformational states, multichain complexes, and inherently disordered regions. For novel folds or complex assemblies, experimental validation is frequently necessary.
Another significant tool is RoseTTAFold, developed by the Baker’s Lab at the University of Washington [71]. RoseTTAFold employs a three-track network model that simultaneously integrates sequence, distance, structure information, allowing for rapid and accurate structure prediction. It is useful substitute for Alphafold 2 because of its design, which permits effective modelling even with limited evolutionary information. ResNet is used to extract evolutionary traits[67] ,GNNs are used to spatial model constraints and folding mechanisms[68] ,and attention- based networks are used to predict inter-residue distances and orientations.
Other emerging DL (https://zhanggroup.org/DeepFold/) ,trRosetta (https://yanglab.qd.sdu.edu.cn/trRosetta/), and RaptorX (https://raptorx.uchicago.edu/StructurePropertyPred/predict/). OmegaFold utilizes protein language model, and does not require MSAs. Making it faster and more effective on for divergent sequences, including those with few homologs [69]. DeepFold is designed for de novo protein structure prediction, using Spatial constraints, which are then assembled into full length models using folding algorithm [70]. Deepfold is de novo PSP tool that uses convolutional residual neural network to predict spatial constraints. Folding algorithms are then used to put the predictions together into full- length models. Homologous templates are incorporated into the network’s predictions to improve accuracy, Rosetta constructs protein structures by direct energy minimization under the inter-residue distance and orientation distributions [71], [72], [73] . RaptorX applies deep convolutional neural fields (DeepCNF) for concurrent prediction of protein structure, solvent accessibility, and disorder regions [74]
While accurate prediction of protein structures forms the foundation for understanding protein function, a critical next step in biomedical research is to elucidate how these proteins interact with small molecules, or ligands. Such protein-ligand interactions underpin most biological processes and are central to drug discovery efforts. Building on advances in protein structure prediction, AI is now increasingly applied to predict and analyze protein-ligand interactions with remarkable accuracy.
Protein ligand interaction prediction
Protein-ligand interaction (PLIs) are fundamental to biological processes and therapeutics development, as they occur when a small molecule (ligand) binds to the target protein (receptor), thereby its function. The strength of this interaction, known as binding affinity, is a key determinant of how effectively a ligand modulates protein activity. Advanced AI and ML algorithms have become powerful tools for predicting and amazing this interaction, accelerating drug discovery and biomedical research.
A variety of curated databases, such as BindingDB, PDBbind, PubChem, and ChEMBL, provide comprehensive data on compound- protein pairs and their corresponding interaction labels[75]. Each molecule is represented using feature vectors or matrices extracted from various biological, topological, and physicochemical properties, which are then used to train ML models [76]. DL architectures, including CNNs, RNNs, GNNs, and transformer-based models, are used to capture complex patterns within these dataset [82]. For instance, CNNs and RNNs are commonly used for ligand binding site prediction, identifying potential pockets on the protein surface where ligands may bind, which is crucial for rational drug design. Notably AI-based ligand binding site prediction tools are P2RANK, a stand-alone template-free tool [78], Deepsite uses 3D CNN [79], GeoNetachieves introduces a coordinate-free geometric representation to characterize local residue distributions and generating an eigenspace to depict local interactive biophysical environments [80].
AI-Driven Virtual Screening
Virtual screening (VS) is an important component of PLIs, leveraging AI models to rapidly screen large libraries of potential drug molecules against target protein. This process help identify candidates with the highest binding affinity, significantly reducing both the time and cost associated with traditional drug development [81]. VS approaches typically classified is structure-based and ligand-based [82].
Structure-based virtual screening (SBVS) requires detailed structural information about the target protein, which can be extracted from experimental approaches such as (NMR) and computational modelling [82]. AI-driven techniques like molecular docking predict how well a drug binds to its target protein, based on its 3D structure. For example, DL-based molecular docking tool utilizes quantitative structure-activity analysis (QSAR) models trained on actual docking scores from a small subset of a molecular database to predict docking scores of the remaining compounds [83], [84]. Advanced tools like DiffDock use a diffusion generative model to sample ligand poses, mapping the manifold of ligand confirmations relevant to degrees of freedom, such as translation, rotation, and torsion [85]. EquiBind, an SE (3)- equivariant geometric DL model can directly predict both the binding location (blind docking) and the bound pose and orientation [86].TankBind, incorporates trigonometry constraints and segments the protein into functional blocks to explicitly attend to all possible binding sites [87]. Uni-Mol utilizes SE (3)-Transformer architecture, with pertaining on extensive molecular and protein pocket datasets, and offers several fine-tuning strategies for downstream tasks [88].SBVS effectively determines PLIs, Key amino acids involved in the interaction, and target’s structural context. ML-based Scoring algorithms, such as NN-score, CS-score, SVR-score, and ID-score, have been developed to further improve prediction accuracy in SBVS.
Ligand-based virtual screening (LBVS), in contrast, relies on the chemical and physiochemical similarities of known compounds to predict new active compounds, without requiring prior structural knowledge of the target protein. AI-driven LBVS models can efficiently identify bioactive molecule using supervised learning on curated datasets of active and inactive compounds. Algorithms such as GNMs and ANNs, as well as models like PARASHIFT, HEX, USR, and ShaPE are commonly employed. After identifying promising compound, further analysis such as ADMET (absorption, distribution, metabolism, excretion, toxicity) profiling and in vitro bioassays were performed, with successful advancement towards clinical trials.
With the progression of AI and ML, several tools for VS have been developed, including ChemSAR[89],Gypsum-DL[90], PyRMD[91]VSFlow, CompScore[92]FlexX-Scan[93],EasyVS[94], MTiOpenScreen[95], Deep Docking[96], RosettaVS[97], A-HIOT[98]. These tools have improved prediction accuracy and reduced false positives, streamlining the drug discovery pipelines. Despite these advances, several challenges remain. The quality and diversity of training data can limit model generalizability, and static predictions may fail to capture dynamic protein-ligand interactions. High-resolution docking and generative modeling are computationally intensive, and experimental validation remains essential to confirm AI-driven predictions.
AI in system biology and multi omics integration
Systems biology takes a holistic approach to understanding biological processes by integrating data from various omics disciplines [99].In contrast to reductionist methodologies focusing on isolated molecular entities, systems biology integrates diverse data sources to construct comprehensive models of biological function and disease mechanisms[99]. Network Analysis is one of the primary methodologies in systems biology, enabling the visualization and interpretation of complex biological interactions. By representing molecular entities as nodes and their interactions as edges, network-based approaches facilitate the identification of main regulatory elements, disease-associated biomarkers, and potential therapeutic targets.[100].The advent of AI has significantly enhanced systems biology and network analysis by enabling efficient processing of large-scale omics data, identifying hidden patterns, and predicting dynamic biological behaviors.
NETWORK INFERENCE
Network Inference is widely applied across various biomedical subfields, such as genomics, metagenomics, epidemiology, and neuroscience [101]. Networks serve as powerful tools for representing complex interactions, from molecular markers and neuronal connections to microbial communities and populations level dynamics [102]. In the context of GRN, network inference aims to reconstruct interaction maps for gene expression data, revealing how genes regulate each other. This is a pivotal tool for understanding complex biological processes and diseases like NDDs and cancer. The goal is to construct a network (graph) where nodes represent elements (e.g., genes, proteins, neurons), and edges represent relationships or interactions between them[103].
Various methods are available for network inference, including correlation-based methods, regression techniques, Bayesian networks, and information-theoretic measures. Each approach has its strengths and weaknesses, some excel at detecting direct interactions, while others perform better suited for capturing non-linear relationships or dynamic regulatory mechanisms. The development of diverse computational tools and algorithms has facilitated network inference. Conventional methods, such as Weighted Gene Co-expression Network Analysis (WGCNA), rely on correlation, while Bayesian network-based approaches infer probabilistic relationships between genes [103]. Advanced algorithms, such as ARACNE (Algorithm for the Reconstruction of Accurate Cellular Networks) and GENIE3, utilize mutual information and tree-based methods to improve accuracy and scalability. [104]. However, newer methods such as Phixer and PIDC (Partial Information Decomposition and Context aim to reduce redundancy and improve directionality in inferred networks [105]. Many of these tools are tested and benchmarked using publicly available datasets such as those from the DREAM Challenges, which provide a standardized framework for evaluating network inference algorithms.
Despite significant progress, several computational challenges remain in network inference. One major issue is the integration of multiple data sources, such as transcriptomics, epigenomics, and proteomics, to improve inference accuracy. Another challenge is the construction of pseudo-temporal orderings from static single-cell RNA sequencing data, which would enable the study of dynamic regulatory interactions. Additionally, combining multiple network inference algorithms has shown promise in improving prediction accuracy, but finding optimal strategies for integration remains an open question. Addressing these challenges will enhance the ability of network inference to generate biologically meaningful insights, particularly in applications such as drug discovery, cancer research, and personalized medicine. Pathway analysis (PA) builds on network-based approaches by identifying functional modules and signaling cascades within these networks, enabling researchers to interpret how groups of genes and proteins work together to drive cellular processes and disease mechanisms which is described in the next section.
| Domain | Tools | Algorithm | Key Features | Limitations | References |
|---|---|---|---|---|---|
| Genome assembly | SPAdes | Hybrid | Improved accuracy in single-cell and bacterial assemblies | Computationally intensive | [38] |
| Seq2squiggle | FFT | Signal prediction faster and more stably | [106] | ||
| Variant calling | Deep Variant | CNN | Enhance genomic analysis accuracy, automatic feature extraction | Computationally intensive and require a large amount of training data | [107] |
| GATK | ML/Stats | Robust, optimized for high-throughput | Complex setup, resource intensive | [108] | |
| Gene expression analysis | XGBoost, RF, k-NN | Classification, feature selection, pattern recognition | High accuracy, handles high-dimensional data | Sensitive to imbalance, batch effects | [109] |
| SVM, GPC | DEG classification, non-linear modeling | Efficient for small/medium datasets | Less interpretable, kernel selection critical | [109] | |
| Autoencoders, CNN | Dimensionality reduction, feature extraction | Captures complex patterns | May lack interpretability | [110] | |
| Transformers | scRNA-seq analysis, rare cell type identification | Improved rare cell detection | Computationally demanding | [111] | |
| Protein structure prediction | Alphafold | Transformer and GNN, MSA based | High accuracy, revolutionized protein modeling | Limited to disordered regions, single conformers only | [112] |
| RoseTTA Fold | Three-track network (ResNet, GNN) | Fast and accurate | Slightly less accurate for large complexes | [113] | |
| OmegaFold | Protein language model | MSA-independent, fast, scalable | Lower resolution for large/divergent proteins | [114] | |
| Protein–ligand interaction prediction | P2RANK, DeepSite | 3D CNN | Template-free, spatial accuracy | Model interpretability, dataset bias | [78] |
| DeepDock, DiffDock | Docking score prediction, diffusion models | Efficient, handles pose flexibility | Relies on the docking dataset quality | [85] | |
| EquiBind, TankBind | SE(3) equivariant, geometric DL | Direct pose prediction, considers protein flexibility | Requires high-quality structures | [86], [87] | |
| ChemSAR, Gypsum-DL | Virtual screening, scaffold generation | High throughput, reduces false positives | Training data bias, chemical space coverage | [89], [90] | |
| Network Analysis | ARACNE, GENIE3 | Mutual information, tree-based inference | Captures nonlinearities, scalable | May infer indirect associations | [104] |
| Phixer, PIDC | Redundancy reduction, directionality improvement | Improved accuracy, directionality | Computational complexity | [105] | |
| Pathway Analysis | DeepGSEA | Prototype-based, scRNA-seq enrichment | Handles heterogeneity, explicit visualizations | Requires high-quality gene sets | [115] |
| PathGNN, PathCNN | GNN/CNN-based, pathway topology integration | Enhanced prediction, interpretable pathways | Needs large annotated datasets | [116] | |
| Dynamic Modeling | dFBA, Cobrapy, CaSQ | Time-dependent metabolic flux simulation, Boolean model | Simulates temporal behaviour | Parameter identifiability, simplifications | [117] |
Table 2: Comparative Overview of Bioinformatics Tools Across Key Application Domains
Pathway Analysis
Pathway analysis (PA) is an essential approach for the identification of significant biological pathways associated with a gene list retrieved from omics datasets[118], [119]. Traditional methods rely on statistical enrichment techniques such as over-representation and gene set enrichment analysis (GSEA)[119]. AI and ML-powered PA, has transformed bioinformatics by enabling the prediction, modelling, and understanding of complex biological interactions. AI-driven PA is crucial for elucidating disease mechanisms, identifying drug targets, predicting drug-drug interactions, and optimizing therapeutic strategies, while facilitating meaningful interpretation and hypothesis generation.
AI-Driven Methodologies in PA ML-based PA approaches include RF, which identifies key pathways based on ranking feature selection from omics data, SVM, enhances GSEA predictions by differentiating relevant vs. non-relevant pathways, and GBM, captures non-linear relationships in gene expression analysis.[120], [121], [122]. DL based PA methods, named as autoencoders, that reduce dimensionality and uncover hidden patterns in gene expression data, GNNs integrate pathway interaction networks based on complex gene interactions, and RNNs capture temporal dependencies in dynamic pathway regulations, especially in time series transcriptomics data. Some commentary techniques such as NLP analyzes biomedical literature to identify novel gene-pathway associations, continuously updating pathway databases with the latest findings. Reinforcement learning algorithms dynamically refine pathway selection strategies, optimizing enrichment scores for improved precision in high-throughput datasets. The following case studies illustrate the diverse and impactful applications of AI-driven approaches in PA PathGNN, GNN model that leverages pathway topology to enhance predictive accuracy in cancer survival prediction. PathGNN outperformed traditional methods by identifying biologically relevant pathways linked to survival outcomes [123] PathCNN, adapts CNNs for pathway-based multi-omics data analysis using innovative pathway image representation. Applied to Glioblastoma multiforme, it predicted long-term survival and identified critical pathways linked to survival outcomes. By improving interpretability and incorporating pathway topology, it enhances the understanding of the underlying biological processes driving disease progression [116] DeepGSEA, improves PA-based GSEA using prototype-based DL for improved interpretability and accuracy in single-cell RNA-seq data. DeepGSEA captures complex gene set patterns and visualizes pathway distributions, aiding in biomarker discovery in precision medicine [115].
MinePath integrates GNNs, network-based scoring methods, and influence propagation models, to uncover regulatory mechanisms (e.g.’CXCR4 mutant gene, ErbB signaling) in breast cancer and cervical cancer datasets. In a study by Yuan et al., the unsupervised DeepT2Vec autoencoder generated 30-dimensional transcriptomic feature vectors (TFV) from 20,000 normal/tumor transcriptomes, while the supervised classifier DeepC achieved to 90% pan-cancer and 94% cancer specific [124].
While PA identifies critical biological pathways and their roles in disease mechanisms, understanding how these pathways evolve over time and respond to perturbations requires dynamic modeling (DM). Dynamic modeling builds on pathway-centric insights by simulating temporal changes in molecular interactions, metabolic fluxes, and signaling cascades. This shift from static pathway mapping to time-resolved simulations enables researchers to predict how biological systems adapt to therapeutic interventions, environmental stresses, or genetic alterations. The following section explores how AI-driven dynamic modeling integrates pathway data, multi-omics inputs, and biophysical constraints to unravel the temporal dynamics of complex biological systems, bridging the gap between functional annotation and predictive systems biology.
Dynamic Modeling
A well-established tool to understand metabolic networks, the temporal behaviour of complex biological systems, such as metabolic pathways, gene regulatory networks, and PPIs, is dynamic modeling (DM) [125]. A key application of DM is in drug response prediction, in which mathematical models simulate how biological systems evolve in response to therapeutic interventions. Dynamic Flux Balance Analysis (dFBA) [117] is a key tool for DM, extending Flux Balance Analysis by incorporating time-dependent constraints.
In metabolic modeling, AI algorithms and bioinformatics tools enhance dFBA. For example, Aghakhani et al. utilized DM to investigate metabolic programming of breast cancer-associated fibroblasts (CAFs) in the tumor microenvironment (TME). They constructed a Boolean model using CaSQ tools to map the regulatory framework, integrating it with MitoCore’s central metabolism network. Flux Balance Analysis (FBA) with CobraPy quantified metabolic fluxes. AI improved biological relevance through enhanced network inference, parameter optimization, and metabolic flux predictions. The study compared two FBA scenarios: a control representing baseline metabolic constraints and a CAF regulatory model. The primary goal was to maximize ATP production, focusing on glycolysis and oxidative phosphorylation (OXPHOS) as key pathways. Since MitoCore lacks tissue specificity, flux distributions (rather than absolute values). ATP production ratios from glycolysis and OXPHOS, alongside carbon uptake/secretion fluxes, revealed metabolic exchanges with the TME. Internal metabolic fluxes comparisons, showed significant alterations, particularly with variations exceeding two-fold Another study leveraged double-hybrid continuous approach to develop a multiscale bioinformatics framework integrating tissue, cellular, and molecular interactions within the TME. This method enables the dynamic simulation of tumor progression and therapeutic response by incorporating vascular networks, metabolic pathways, and drug diffusion models. By treating tumor vasculature and drug distribution as interconnected tissues, the model captures the spatiotemporal evolution of tumor heterogeneity. The study highlights the importance of network modeling in predicting combination therapy efficacy, optimizing metronomic chemotherapy, and improving drug penetration through vascular normalization strategies[127].
Rachel et al extended the Retarded Transient Function approach to model both temporal and dose dependent dynamics in intracellular signaling networks. This method provides a computationally efficient and interpretable framework for predicting of signaling differences across biological conditions in their response to stimuli. Using Inflammasome activation in bone marrow-derived macrophages as a case study, the model successfully characterized dependencies, dose-response kinetics, and signaling dynamics [128].
Wang et al developed a multiscale in silico model integrating EGFR-ERK signaling and cellular dynamics in Non-Small Cell Lung Cancer (NSCLC). The model demonstrates how extrinsic ligand concentrations and intrinsic molecular profiles influence tumor spatial dynamics, revealing a phase transition where a minimal ligand increase suppresses proliferation. These findings highlight the importance of feedback mechanisms between molecular and cellular scales in shaping tumor behavior[129].
Challenges and limitations of AI in bioinformatics
Data Quality and Availability
The performance and accuracy of AI-driven bioinformatics applications heavily depend on the quality and availability of biological data. While high-throughput sequencing and omics technologies generate vast amounts of data, these datasets often suffer from noise, missing values, and inconsistencies [125]. The lack of standardized formats and integration across databases further complicates data accessibility and usability. Additionally, data privacy regulations and ethical considerations restrict access to patient-derived genomic and clinical datasets, limiting the scope of AI applications in precision medicine.
AI models require diverse and representative datasets for robust training and generalization. However, biases in available datasets can lead to skewed predictions, reducing the reliability of AI-driven insights [126]. Addressing data quality issues through improved curation, annotation, and harmonization strategies is essential for enhancing the effectiveness of AI in bioinformatics.
Reproducibility and Validation of AI Results
Reproducibility is a critical issue in AI-driven bioinformatics research. AI models often rely on complex computational pipelines, sensitive to variations in dataset preprocessing, algorithm selection, and hyperparameter tuning [130]. Differences in computational environments and software dependencies can lead to inconsistent results, making it challenging to validate AI findings across different research groups.
To address this challenge, researchers emphasize the importance of open-source tools, standardized benchmarking datasets, and transparent reporting of methodologies. Reproducibility initiatives, such as FAIR (Findable, Accessible, Interoperable, and Reusable) data principles and AI model repositories, play a crucial role in improving reliability and facilitating independent validation of AI-based bioinformatics studies [131], [132].
Integration with Experimental Methods
Despite its computational capabilities, AI in bioinformatics must be effectively integrated with experimental methods to provide meaningful biological insights. AI models generate predictions that require validation through laboratory techniques like CRISPR gene editing, mass spectrometry, and high-throughput screening [129]. The continuous feedback loop between AI predictions and experimental results is vital for refining models and enhancing their biological relevance.
However, the integration of AI with experimental workflows poses challenges, particularly in fostering effective collaboration between computational scientists and experimental biologists. The lack of standardized approaches for validating AI-generated hypothesis highlights the need of strong frameworks to support AI driven discovery and its translation into practical applications.
As the field continues to address these foundational challenges, attention is increasingly turning to the future directions and transformative opportunities that AI offers in bioinformatics research.
Future of AI in bioinformatics
Integration of AI in Bioinformatics
The integration of AI into bioinformatics is poised to dramatically change the nature of biotechnology research. Recent advances in AI, coupled with breakthroughs in ML, robotics, and data analytics, display enormous potential to revolutionize the field in ways once thought unimaginable. However, these advancements, also raise significant ethical, labor, and security challenges that must be carefully addressed. Serving mankind ethically requires ensuring fair access to AI’s advantages while reducing its hazards. Among the emerging priorities in AI integration is the need for transparency and interpretability, particularly as AI models become more complex and widely adopted in sensitive domains such as healthcare.
Explainable AI (XAI) in Bioinformatics
The "Black box" problem remains a significant challenge in AI-driven bioinformatics, where conventional AI models lack transparency, obscuring how input data are transformed into output results[133]. For example, in an ANN, contains multiple interconnected layers, such as input, hidden, and output layers, with hidden layers, posing interpretability challenges due to their complex internal structure. The intricate internal structure obscures the reasoning behind specific predictions and decisions.
Explainable AI (XAI) has emerged as a critical solution to this issue. XAI techniques open the black box by providing insight s into how model operate, thereby improving both the predictability and trustworthiness of AI systems [134]. XAI works by analyzing the influence of each feature on the model's behavior. In addition to model visualization, methods such as saliency maps, feature importance ranking, and decision trees are used to clarify the model's decision-making process. These methods help researchers and clinicians understand, interpret, and trust model outouts by revealing the factors driving predictions.
Improving interpretability through XAI, not only enables the identification and reduction of biases but also to establish confidence in AI-driven results. In bioinformatics, where datasets, are often large complex, and heterogenous interpretability is crucial. Without it, the basis for model predictions can be lost amid the complexity of the data and algorithms. XAI provides a valuable toolkit for healthcare professional to validate, optimize, and deploy AI models more reliably in clinical and research settings [135], [136]. XAI technologies deployed in various fields, like predictive modeling, data interpretation, and mining valuable patterns from unstructured data in the biological domain. For example, interpretable DL methods such as SHapley Additive exPlanations (SHAP) and class activation maps (grad-CAM) have been proven effective for analyzing DNA, RNA, and protein sequences [136], [137]. Similarly, tools like Local Interpretable Model-agnostic Explanations (LIME) have become more popular in bioimaging, including CT and MRI image assessments. LIME segments images into interpretable "superpixels," making it possible to quantify and visualize region of interest, leading to more accurate identification of the disease and improved diagnostic results[135].
In summary, XAI is important to bridge the gap between complex AI models and practical trustworthy applications in bioinformatics. By improving transparency and interpretability, XAI empowers to make better informed decisions, ultimately improving the reliability and impact of AI- driven discoveries in biomedical sciences.
Transfer Learning and Domain Adaptation
The performance of AI models in bioinformatics, particularly in applications like bioimaging, depends heavily on the quality and consistency of the training data [138]. If the dataset is heterogeneous and unbiased, generally the model will provide accurate results, while a flawed or biased dataset can significantly complicate the accurate assessment of the model performance.
Transfer learning allows model trained on one task to be repurposed for related tasks, leveraging prior knowledge to improve performance [139], [140].This approach facilitates bias detection, model validation and efficient resource utilisation. By redefining the data properties to achieve domain invariance, domain adaptation ensures models remain robust across varied biological context[141]. These methodologies are becoming important in bioinformatics applications, such as clinical image analysis, tissue segmentation, disease classification, and gene expression profiling, where they enhance model accuracy and generalizability[142].
Federated Learning
As AI expands sensitive domains like healthcare, important ensuring data privacy and security becomes paramount. Federated learning offers a decentralized approach, allowing organizations to collaboratively train AI models without sharing raw data [143], [144].Only the trained model is exchanged, preserving confidentiality and reducing the risk of data breaches.
This approach helps organisations to train AI models on their own datasets without the risk of data transfer, thereby offering increased security. The model that is only to be shared is the trained one, which means the raw data stays safe and confidential. This technique not only averts data loss incidents but also [145], [146]. Federated learning is especially beneficial in the case of large-scale, multi-center genomic studies. The genomic information has always been the most sensitive among all the biological information because federated learning here can enable the research centres to work together on predictive models, e.g., for disease risk assessment or pharmacogenomic responses, without the necessity of raw genomic data gathering, and thus, data privacy is preserved while the innovation is encouraged [145], [146].
Quantum Computing and Next-Generation AI
Quantum computing represents a new era in computational power, that can deliver certain computing tasks and data storage at levels far higher than those of ordinary computers. In contrast to quantum computers, where information is typically represented as binary bits (0s and 1s), ordinary computers are resource-consuming to process large datasets. Quantum computers mainly rely on qubits, taking advantage of the concept of superposition, to exist in different states at the same time. This core contrast is responsible for the fact that quantum computers can solve complicated problems and handle high-dimensional data sets very quickly and with good accuracy [147].
In bioinformatics, quantum computing can significantly advance several of the most challenging and difficult tasks in this field like analyzing the massive amount of biological data, modeling molecular interactions, and simulating biological systems without any inaccuracies. These capabilities could accelerate drug discovery, and genomics research and therapeutics development, drastically shortening timelines accuracy [148].
Digital Twin Technologies in Healthcare
Digital twin technology is an emerging in-silico method, with significant potential in healthcare, with its approach to model and track patient health data in a live mode being the state-of-the-art method. A digital twin is dynamic, virtual representation of a physical system-such as a patient-created by integrating data medical imaging, wearables sensors, genomes, and clinical records. This technology makes real-time simulation and monitoring of a person's health status, supporting, personalized diagnosis and treatment [149].
Creating a Digital Twin Involves Several Stages:
- Data Acquisition
- Medical Imaging (e.g., MRI, CT, Ultrasound):
Researchers used admitted patient's body images to create a geometrically accurate model, establishing a new standard in the healthcare sector [150]. - Wearable Sensor Data:
Data on physical parameters such as heart rate, glucose, and sleep were monitored in real-time using body-worn devices. Bruynseels et al. identified this as the next step in digital care [150]. - Omics Data (Genomics, Proteomics, Metabolomics):
Omics technology plays a pivotal role in identifying genetic predispositions, molecular pathways, and biomarkers. The new era of computational systems biology and functional genomics maximizes the potential of these discoveries [150]. - Clinical Records:
Electronic health records serve as comprehensive repositories of patient histories, diagnoses, and more, maintained by hospitals. This CBMM approach aims to accelerate patient diagnosis and assist healthcare providers in making optimal decisions [149].
- Medical Imaging (e.g., MRI, CT, Ultrasound):
- Model Integration
- Computational Modelling:
Simulates organ and tissue behaviour using systems biology and agent-based modeling, providing detailed representations of physiological processes and interactions [150]. - ML & AI:
Detects complex patterns within diverse datasets, enhancing the accuracy and precision of predictions to support personalized diagnostics and treatment planning [151].
- Computational Modelling:
- Continuous Updating
- Feedback Loops:
Real-time patient data is continuously incorporated into the model, ensuring that the simulations remain up-to-date and reflective of the patient’s evolving health status [152]. - Predictive Algorithms:
These algorithms dynamically generate and refine diagnostic and treatment strategies, allowing the digital twin to serve as a living, adaptive model of the patient’s healthcare [153].
- Feedback Loops:
A digital twin thus provides medical professionals with a comprehensive real-time view of the patient with minimal invasiveness. This approach enables more accurate diagnosis, increases the possibility of high-quality treatment, and empowers patients with detailed health records to make informed decisions regarding their care [154], [155].
Conclusion
AI, encompassing ML and DL, is a powerful computational technique that has already transformed several areas of research. With the recent explosion of genetic, molecular, and clinical data, ML provides novel techniques for interrogating, analysing, and processing this data, as well as extracting significant new knowledge about the underlying processes. ML techniques are particularly appealing in computational biology because of their capacity to rapidly produce predictive models in the absence of strong assumptions about the underlying mechanisms, which is typical of some of biomedicine's most serious concerns. From genome assembly and variant calling to proteomics research, gene expression analysis, and drug development, DL techniques have demonstrated impressive effectiveness. They frequently outperform conventional computer techniques in terms of accuracy and scalability. However, several challenges still exist. For AI-driven bioinformatics to be reliable, a variety of high-quality, well-annotated datasets must be available. Issues such as noise, biases, and heterogeneous data can pose challenges to the universality and performance of the model. The interpretability of DL models remains a significant challenge since many state-of-the-art architectures function as “black boxes” , limiting transparency and confidence in crucial biological applications. Furthermore, reproducibility and standardization of AI procedures are essential to ensure that computational results are converted into reliable biological insights and therapeutic effects. Numerous significant elements will impact bioinformatics advancements in the future. Integrating multi-omics data, developing interpretable and explicable AI models, utilizing transfer learning and domain adaptation, and putting privacy-preserving techniques like federated learning into practice are all crucial for the development of computers. Emerging technologies like digital twin systems and quantum computing have the potential to speed up research and make preventive, predictive, and customized healthcare possible. To achieve these goals, interdisciplinary collaboration, careful benchmarking, and a commitment to ethical and responsible AI deployment are required.
Authors contribution
AKM, SS, NR, PC, UK, and B conceived and designed this project. Data collected and analyzed by NR, PC, UK, N, NK, and KK. Manuscript have been written by NR, PC, B, UK, N, KK, NK, AKM, and SS.
Conflict of Interest
The authors declare no conflict of interest
Acknowledgment
We would like to thank senior management of Delhi Technological University (DTU), Delhi University (DU), University Grant commission (UGC), and Council for Scientific and Industrial Research (CSIR).
References
- A. Bayat, “Science, medicine, and the future: Bioinformatics,” BMJ, vol. 324, no. 7344, pp. 1018–1022, Apr. 2002, doi: 10.1136/bmj.324.7344.1018.
- K. Nakamae and H. Bono, “Genome editing and bioinformatics,” Gene and Genome Editing, vol. 3–4, p. 100018, Dec. 2022, doi: 10.1016/j.ggedit.2022.100018.
- T. T. Ogunjobi et al., “Bioinformatics Applications in Chronic Diseases: A Comprehensive Review of Genomic, Transcriptomics, Proteomic, Metabolomics, and Machine Learning Approaches,” Medinformatics, Feb. 2024, doi: 10.47852/bonviewMEDIN42022335.
- A. Bin Rashid and M. A. K. Kausik, “AI revolutionizing industries worldwide: A comprehensive overview of its diverse applications,” Hybrid Advances, vol. 7, p. 100277, Dec. 2024, doi: 10.1016/j.hybadv.2024.100277.
- C. Campbell, “Machine Learning Methodology in Bioinformatics,” in Springer Handbook of Bio-/Neuroinformatics, Berlin, Heidelberg: Springer Berlin Heidelberg, 2014, pp. 185–206. doi: 10.1007/978-3-642-30574-0_12.
- M. A. Khan, R. Khan, F. Algarni, I. Kumar, A. Choudhary, and A. Srivastava, “Performance evaluation of regression models for COVID-19: A statistical and predictive perspective,” Ain Shams Engineering Journal, vol. 13, no. 2, p. 101574, Mar. 2022, doi: 10.1016/j.asej.2021.08.016.
- K. Lee et al., “Scaling up data curation using deep learning: An application to literature triage in genomic variation resources,” PLoS Comput Biol, vol. 14, no. 8, p. e1006390, Aug. 2018, doi: 10.1371/journal.pcbi.1006390.
- A. Monaco et al., “A primer on machine learning techniques for genomic applications,” Comput Struct Biotechnol J, vol. 19, pp. 4345–4359, 2021, doi: 10.1016/j.csbj.2021.07.021.
- M. D. Saçar Demirci, J. Baumbach, and J. Allmer, “On the performance of pre-microRNA detection algorithms,” Nat Commun, vol. 8, no. 1, p. 330, Aug. 2017, doi: 10.1038/s41467-017-00403-z.
- J. Kim, V. D. Calhoun, E. Shim, and J.-H. Lee, “Deep neural network with weight sparsity control and pre-training extracts hierarchical features and enhances classification performance: Evidence from whole-brain resting-state functional connectivity patterns of schizophrenia,” Neuroimage, vol. 124, pp. 127–146, Jan. 2016, doi: 10.1016/j.neuroimage.2015.05.018.
- M. Spencer, J. Eickholt, and J. Cheng, “A Deep Learning Network Approach to <italic>ab initio</italic> Protein Secondary Structure Prediction,” IEEE/ACM Trans Comput Biol Bioinform, vol. 12, no. 1, pp. 103–112, Jan. 2015, doi: 10.1109/TCBB.2014.2343960.
- R. Heffernan et al., “Improving prediction of secondary structure, local backbone angles and solvent accessible surface area of proteins by iterative deep learning,” Sci Rep, vol. 5, no. 1, p. 11476, Jun. 2015, doi: 10.1038/srep11476.
- Y. Chen, Y. Li, R. Narayan, A. Subramanian, and X. Xie, “Gene expression inference with deep learning,” Bioinformatics, vol. 32, no. 12, pp. 1832–1839, Jun. 2016, doi: 10.1093/bioinformatics/btw074.
- R.’ ’Fakoor, F. ’Ladhak, A. ’Nazi, and “Huber.Manfred,” “Using deep learning to enhance cancer diagnosis and classification,” in The 30th International Conference on Machine Learning (ICML 2013),WHEALTH workshop, 2013.
- B. Alipanahi, A. Delong, M. T. Weirauch, and B. J. Frey, “Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning,” Nat Biotechnol, vol. 33, no. 8, pp. 831–838, Aug. 2015, doi: 10.1038/nbt.3300.
- H. Zeng, M. D. Edwards, G. Liu, and D. K. Gifford, “Convolutional neural network architectures for predicting DNA–protein binding,” Bioinformatics, vol. 32, no. 12, pp. i121–i127, Jun. 2016, doi: 10.1093/bioinformatics/btw255.
- J. Zhou and O. G. Troyanskaya, “Predicting effects of noncoding variants with deep learning–based sequence model,” Nat Methods, vol. 12, no. 10, pp. 931–934, Oct. 2015, doi: 10.1038/nmeth.3547.
- S. K. Sønderby, C. K. Sønderby, H. Nielsen, and O. Winther, “Convolutional LSTM Networks for Subcellular Localization of Proteins,” 2015, pp. 68–80. doi: 10.1007/978-3-319-21233-3_6.
- B. Lee, J. Baek, S. Park, and S. Yoon, “deepTarget: End-to-end Learning Framework for microRNA Target Prediction using Deep Recurrent Neural Networks,” Mar. 2016.
- S. Min, B. Lee, and S. Yoon, “Deep learning in bioinformatics,” Brief Bioinform, p. bbw068, Jul. 2016, doi: 10.1093/bib/bbw068.
- T. Mikolov, K. Chen, G. Corrado, and J. Dean, “Efficient Estimation of Word Representations in Vector Space,” Jan. 2013.
- A. Vaswani et al., “Attention Is All You Need,” Jun. 2017.
- N. M. Rezk, M. Purnaprajna, T. Nordström, and Z. Ul-Abdin, “Recurrent Neural Networks: An Embedded Computing Perspective,” Jul. 2019, doi: 10.1109/ACCESS.2020.2982416.
- A. Miltiadous, E. Gionanidis, K. D. Tzimourta, N. Giannakeas, and A. T. Tzallas, “DICE-Net: A Novel Convolution-Transformer Architecture for Alzheimer Detection in EEG Signals,” IEEE Access, vol. 11, pp. 71840–71858, 2023, doi: 10.1109/ACCESS.2023.3294618.
- E. D. Oikonomou, P. Karvelis, N. Giannakeas, A. Vrachatis, E. Glavas, and A. T. Tzallas, “How natural language processing derived techniques are used on biological data: a systematic review,” Network Modeling Analysis in Health Informatics and Bioinformatics, vol. 13, no. 1, p. 23, May 2024, doi: 10.1007/s13721-024-00458-1.
- I. Bianconi, R. Aschbacher, and E. Pagani, “Current Uses and Future Perspectives of Genomic Technologies in Clinical Microbiology,” Antibiotics 2023, Vol. 12, Page 1580, vol. 12, no. 11, p. 1580, Oct. 2023, doi: 10.3390/ANTIBIOTICS12111580.
- J. Zhang, “What Has Genomics Taught An Evolutionary Biologist?,” Genomics Proteomics Bioinformatics, vol. 21, no. 1, p. 1, Feb. 2023, doi: 10.1016/J.GPB.2023.01.005.
- H. Satam et al., “Next-Generation Sequencing Technology: Current Trends and Advancements,” Biology (Basel), vol. 12, no. 7, p. 997, Jul. 2023, doi: 10.3390/BIOLOGY12070997.
- S. Brasil et al., “Artificial Intelligence in Epigenetic Studies: Shedding Light on Rare Diseases,” Front Mol Biosci, vol. 8, p. 648012, May 2021, doi: 10.3389/FMOLB.2021.648012/BIBTEX.
- R. Prabhakaran et al., “Epigenetic frontiers: miRNAs, long non-coding RNAs and nanomaterials are pioneering to cancer therapy,” Epigenetics & Chromatin 2024 17:1, vol. 17, no. 1, pp. 1–26, Oct. 2024, doi: 10.1186/S13072-024-00554-6.
- A. M. Giani, G. R. Gallo, L. Gianfranceschi, and G. Formenti, “Long walk to genomics: History and current approaches to genome sequencing and assembly,” Comput Struct Biotechnol J, vol. 18, pp. 9–19, Jan. 2020, doi: 10.1016/J.CSBJ.2019.11.002.
- S. Schmeing and M. D. Robinson, “Gapless provides combined scaffolding, gap filling, and assembly correction with long reads,” Life Sci Alliance, vol. 6, no. 7, p. e202201471, Jul. 2023, doi: 10.26508/LSA.202201471.
- A. R. Soares, P. M. Pereira, and M. A. S. Santos, “Next-generation sequencing of miRNAs with Roche 454 GS-FLX technology: steps for a successful application,” Methods Mol Biol, vol. 822, pp. 189–204, 2012, doi: 10.1007/978-1-61779-427-8_13.
- S. Bhaskaran and C. Saikumar, “A Review of Next Generation Sequencing Methods and its Applications in Laboratory Diagnosis,” J Pure Appl Microbiol, vol. 16, no. 2, pp. 825–833, Jun. 2022, doi: 10.22207/JPAM.16.2.45.
- R. P. Baptista et al., “Assembly of highly repetitive genomes using short reads: the genome of discrete typing unit III Trypanosoma cruzi strain 231,” Microb Genom, vol. 4, no. 4, p. e000156, Apr. 2018, doi: 10.1099/MGEN.0.000156.
- V. Peona et al., “Identifying the causes and consequences of assembly gaps using a multiplatform genome assembly of a bird‐of‐paradise,” Mol Ecol Resour, vol. 21, no. 1, p. 263, Jan. 2020, doi: 10.1111/1755-0998.13252.
- D. Beslic, M. Kucklick, S. Engelmann, S. Fuchs, B. Y. Renard, and N. Koerber, “End-to-end simulation of nanopore sequencing signals with feed-forward transformers,” bioRxiv, p. 2024.08.12.607296, Aug. 2024, doi: 10.1101/2024.08.12.607296.
- A. Prjibelski, D. Antipov, D. Meleshko, A. Lapidus, and A. Korobeynikov, “Using SPAdes De Novo Assembler,” Curr Protoc Bioinformatics, vol. 70, no. 1, p. e102, Jun. 2020, doi: 10.1002/CPBI.102.
- D. C. Koboldt, “Best practices for variant calling in clinical sequencing,” Genome Medicine 2020 12:1, vol. 12, no. 1, pp. 1–13, Oct. 2020, doi: 10.1186/S13073-020-00791-W.
- C. Scarano, I. Veneruso, R. R. De Simone, G. Di Bonito, A. Secondino, and V. D’Argenio, “The Third-Generation Sequencing Challenge: Novel Insights for the Omic Sciences,” Biomolecules, vol. 14, no. 5, p. 568, May 2024, doi: 10.3390/BIOM14050568.
- M. Jain, H. E. Olsen, B. Paten, and M. Akeson, “The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community,” Genome Biology 2016 17:1, vol. 17, no. 1, pp. 1–11, Nov. 2016, doi: 10.1186/S13059-016-1103-0.
- I. Henarejos-Castillo et al., “Machine learning-based approach highlights the use of a genomic variant profile for precision medicine in ovarian failure,” J Pers Med, vol. 11, no. 7, Jul. 2021, doi: 10.3390/JPM11070609.
- A. Gmeiner, P. M. K. Njage, L. T. Hansen, F. M. Aarestrup, and P. Leekitcharoenphon, “Predicting Listeria monocytogenes virulence potential using whole genome sequencing and machine learning,” Int J Food Microbiol, vol. 410, Jan. 2024, doi: 10.1016/J.IJFOODMICRO.2023.110491.
- K. P. Singh, C. Miaskowski, A. A. Dhruva, E. Flowers, and K. M. Kober, “Mechanisms and Measurement of Changes in Gene Expression,” Biol Res Nurs, vol. 20, no. 4, p. 369, Jul. 2018, doi: 10.1177/1099800418772161.
- L. Feng et al., “Screening, identification and targeted intervention of necroptotic biomarkers of asthma,” Biochem Biophys Res Commun, vol. 735, Nov. 2024, doi: 10.1016/j.bbrc.2024.150674.
- M. Abadi et al., “TensorFlow: A system for large-scale machine learning,” Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation, OSDI 2016, pp. 265–283, 2016.
- I. Sutskever, O. Vinyals, and Q. V. Le, “Sequence to sequence learning with neural networks,” Adv Neural Inf Process Syst, vol. 4, no. January, pp. 3104–3112, 2014.
- H. Salehinejad, S. Sankar, J. Barfett, E. Colak, and S. Valaee, “Recent Advances in Recurrent Neural Networks,” Dec. 2017, Accessed: Feb. 28, 2025. [Online]. Available: http://arxiv.org/abs/1801.01078
- A. Saxena, “An Introduction to Convolutional Neural Networks,” Int J Res Appl Sci Eng Technol, vol. 10, no. 12, pp. 943–947, Dec. 2022, doi: 10.22214/ijraset.2022.47789.
- Y. Cheng, S. M. Xu, K. Santucci, G. Lindner, and M. Janitz, “Machine learning and related approaches in transcriptomics,” Biochem Biophys Res Commun, vol. 724, p. 150225, Sep. 2024, doi: 10.1016/J.BBRC.2024.150225.
- H. Huang, Y. Wang, C. Rudin, and E. P. Browne, “Towards a comprehensive evaluation of dimension reduction methods for transcriptomic data visualization,” Communications Biology 2022 5:1, vol. 5, no. 1, pp. 1–11, Jul. 2022, doi: 10.1038/s42003-022-03628-x.
- R. Shashikant, U. Chaskar, L. Phadke, and C. Patil, “Gaussian process-based kernel as a diagnostic model for prediction of type 2 diabetes mellitus risk using non-linear heart rate variability features,” Biomed Eng Lett, vol. 11, no. 3, p. 273, Aug. 2021, doi: 10.1007/S13534-021-00196-7.
- Y. Wang et al., “scGREAT: Transformer-based deep-language model for gene regulatory network inference from single-cell transcriptomics,” iScience, vol. 27, no. 4, p. 109352, Apr. 2024, doi: 10.1016/J.ISCI.2024.109352.
- P. Joshi and R. Dhar, “EpICC: A Bayesian neural network model with uncertainty correction for a more accurate classification of cancer,” Sci Rep, vol. 12, no. 1, p. 14628, Aug. 2022, doi: 10.1038/s41598-022-18874-6.
- E. Amiri Souri, A. Chenoweth, A. Cheung, S. N. Karagiannis, and S. Tsoka, “Cancer Grade Model: a multi-gene machine learning-based risk classification for improving prognosis in breast cancer,” Br J Cancer, vol. 125, no. 5, pp. 748–758, Aug. 2021, doi: 10.1038/s41416-021-01455-1.
- C.-H. Lin, S.-I. Chiu, T.-F. Chen, J.-S. R. Jang, and M.-J. Chiu, “Classifications of Neurodegenerative Disorders Using a Multiplex Blood Biomarkers-Based Machine Learning Model,” Int J Mol Sci, vol. 21, no. 18, p. 6914, Sep. 2020, doi: 10.3390/ijms21186914.
- S. N. A. Shah and R. Parveen, “Differential gene expression analysis and machine learning identified structural, TFs, cytokine and glycoproteins, including SOX2, TOP2A, SPP1, COL1A1, and TIMP1 as potential drivers of lung cancer,” Biomarkers, pp. 1–16, Feb. 2025, doi: 10.1080/1354750X.2025.2461698.
- D. Schaack, M. A. Weigand, and F. Uhle, “Comparison of machine-learning methodologies for accurate diagnosis of sepsis using microarray gene expression data,” PLoS One, vol. 16, no. 5, p. e0251800, May 2021, doi: 10.1371/journal.pone.0251800.
- S. Al-Amrani et al., “Proteomics: Concepts and applications in human medicine,” World J Biol Chem, vol. 12, no. 5, p. 57, Sep. 2021, doi: 10.4331/WJBC.V12.I5.57.
- H. V. CR Jimenez, “Mass spectrometry-based proteomics: from cancer biology to protein biomarkers, drug targets, and clinical applications,” Am Soc Clin Oncol Educational Book, vol. 34, no. 1, pp. e504-10, 2014.
- E. Khodadadi, “Proteomic applications in antimicrobial resistance and clinical microbiology studies,” Infect Drug Resist, vol. 13, p. 1785, 2020.
- H. W. Wang and J. W. Wang, “How cryo‐electron microscopy and X‐ray crystallography complement each other,” Protein Sci, vol. 26, no. 1, p. 32, Jan. 2016, doi: 10.1002/PRO.3022.
- J. Wang et al., “Exploring Human Diseases and Biological Mechanisms by Protein Structure Prediction and Modeling,” Adv Exp Med Biol, vol. 939, p. 39, Nov. 2016, doi: 10.1007/978-981-10-1503-8_3.
- R. Nussinov, M. Zhang, Y. Liu, and H. Jang, “AlphaFold, Artificial Intelligence (AI), and Allostery,” J Phys Chem B, vol. 126, no. 34, p. 6372, Sep. 2022, doi: 10.1021/ACS.JPCB.2C04346.
- J. Jumper et al., “Highly accurate protein structure prediction with AlphaFold,” Nature 2021 596:7873, vol. 596, no. 7873, pp. 583–589, Jul. 2021, doi: 10.1038/s41586-021-03819-2.
- M. Baek et al., “Accurate prediction of protein structures and interactions using a three-track neural network,” Science (1979), vol. 373, no. 6557, pp. 871–876, Aug. 2021, doi: 10.1126/SCIENCE.ABJ8754/SUPPL_FILE/ABJ8754_MDAR_REPRODUCIBILITY_CHECKLIST.PDF.
- S. Kaushik, A. G. Nair, E. Mutt, H. P. Subramanian, and R. Sowdhamini, “Rapid and enhanced remote homology detection by cascading hidden Markov model searches in sequence space,” Bioinformatics, vol. 32, no. 3, pp. 338–344, Feb. 2016, doi: 10.1093/BIOINFORMATICS/BTV538.
- D. Lee, D. Xiong, S. Wierbowski, L. Li, S. Liang, and H. Yu, “Deep learning methods for 3D structural proteome and interactome modeling,” Curr Opin Struct Biol, vol. 73, Apr. 2022, doi: 10.1016/J.SBI.2022.102329.
- R. Wu et al., “High-resolution de novo structure prediction from primary sequence,” bioRxiv, p. 2022.07.21.500999, Jul. 2022, doi: 10.1101/2022.07.21.500999.
- J. W. Lee et al., “DeepFold: enhancing protein structure prediction through optimized loss functions, improved template features, and re-optimized energy function,” Bioinformatics, vol. 39, no. 12, Dec. 2023, doi: 10.1093/BIOINFORMATICS/BTAD712.
- J. Michels et al., “Natural Language Processing Methods for the Study of Protein–Ligand Interactions,” J Chem Inf Model, vol. 65, no. 5, pp. 2191–2213, Mar. 2025, doi: 10.1021/acs.jcim.4c01907.
- W. Bao, Y. Cao, Y. Yang, H. Che, J. Huang, and S. Wen, “Data-driven stock forecasting models based on neural networks: A review,” Information Fusion, vol. 113, p. 102616, Jan. 2025, doi: 10.1016/j.inffus.2024.102616.
- R. Krivák and D. Hoksza, “P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure,” J Cheminform, vol. 10, no. 1, p. 39, Dec. 2018, doi: 10.1186/s13321-018-0285-8.
- S. Wang, W. Li, S. Liu, and J. Xu, “RaptorX-Property: a web server for protein structure property prediction,” Nucleic Acids Res, vol. 44, no. Web Server issue, p. W430, Jul. 2016, doi: 10.1093/NAR/GKW306.
- G. A. Abdelkader and J.-D. Kim, “Advances in Protein-Ligand Binding Affinity Prediction via Deep Learning: A Comprehensive Study of Datasets, Data Preprocessing Techniques, and Model Architectures,” Curr Drug Targets, vol. 25, no. 15, pp. 1041–1065, Jan. 2024, doi: 10.2174/0113894501330963240905083020.
- J. Michels et al., “Natural Language Processing Methods for the Study of Protein–Ligand Interactions,” J Chem Inf Model, Feb. 2025, doi: 10.1021/ACS.JCIM.4C01907.
- W. Bao, Y. Cao, Y. Yang, H. Che, J. Huang, and S. Wen, “Data-driven stock forecasting models based on neural networks: A review,” Information Fusion, vol. 113, p. 102616, Jan. 2025, doi: 10.1016/J.INFFUS.2024.102616.
- R. Krivák and D. Hoksza, “P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure,” J Cheminform, vol. 10, no. 1, p. 39, Dec. 2018, doi: 10.1186/s13321-018-0285-8.
- R. Aggarwal, A. Gupta, V. Chelur, C. V. Jawahar, and U. D. Priyakumar, “DeepPocket: Ligand Binding Site Detection and Segmentation using 3D Convolutional Neural Networks,” J Chem Inf Model, vol. 62, no. 21, pp. 5069–5079, Nov. 2022, doi: 10.1021/acs.jcim.1c00799.
- J. Han, S. Zhang, M. Guan, Q. Li, X. Gao, and J. Liu, “GeoNet enables the accurate prediction of protein-ligand binding sites through interpretable geometric deep learning,” Structure, vol. 32, no. 12, pp. 2435-2448.e5, Dec. 2024, doi: 10.1016/j.str.2024.10.011.
- A. I. Visan and I. Negut, “Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery,” Life, vol. 14, no. 2, p. 233, Feb. 2024, doi: 10.3390/LIFE14020233.
- J. Dai et al., “Combined usage of ligand- and structure-based virtual screening in the artificial intelligence era,” Eur J Med Chem, vol. 283, p. 117162, Feb. 2025, doi: 10.1016/J.EJMECH.2024.117162.
- M. T. Muhammed and E. Aki-Yalcin, “Molecular Docking: Principles, Advances, and Its Applications in Drug Discovery,” Lett Drug Des Discov, vol. 21, no. 3, pp. 480–495, Mar. 2024, doi: 10.2174/1570180819666220922103109.
- J. Mao et al., “Comprehensive strategies of machine-learning-based quantitative structure-activity relationship models,” iScience, vol. 24, no. 9, p. 103052, Sep. 2021, doi: 10.1016/j.isci.2021.103052.
- G. Corso, H. Stärk, B. Jing, R. Barzilay, and T. Jaakkola, “DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking,” 11th International Conference on Learning Representations, ICLR 2023, Oct. 2022, Accessed: Feb. 27, 2025. [Online]. Available: https://arxiv.org/abs/2210.01776v2
- H. Stärk, O. E. Ganea, L. Pattanaik, R. Barzilay, and T. Jaakkola, “EquiBind: Geometric Deep Learning for Drug Binding Structure Prediction,” Proc Mach Learn Res, vol. 162, pp. 20503–20521, Feb. 2022, Accessed: Feb. 27, 2025. [Online]. Available: https://arxiv.org/abs/2202.05146v4
- W. Lu, Q. Wu, J. Zhang, J. Rao, C. Li, and S. Zheng, “TANKBind: Trigonometry-Aware Neural NetworKs for Drug-Protein Binding Structure Prediction,” Jun. 06, 2022. doi: 10.1101/2022.06.06.495043.
- G. Zhou et al., “Uni-Mol: A Universal 3D Molecular Representation Learning Framework,” Mar. 07, 2023. doi: 10.26434/chemrxiv-2022-jjm0j-v4.
- J. Dong et al., “ChemSAR: An online pipelining platform for molecular SAR modeling,” J Cheminform, vol. 9, no. 1, pp. 1–13, May 2017, doi: 10.1186/S13321-017-0215-1/TABLES/4.
- P. J. Ropp et al., “GypSUm-DL: An open-source program for preparing small-molecule libraries for structure-based virtual screening,” J Cheminform, vol. 11, no. 1, pp. 1–13, May 2019, doi: 10.1186/S13321-019-0358-3/FIGURES/6.
- G. Amendola and S. Cosconati, “PyRMD: A New Fully Automated AI-Powered Ligand-Based Virtual Screening Tool,” J Chem Inf Model, vol. 61, no. 8, pp. 3835–3845, Aug. 2021, doi: 10.1021/acs.jcim.1c00653.
- Y. Perez-Castillo et al., “CompScore: Boosting Structure-Based Virtual Screening Performance by Incorporating Docking Scoring Function Components into Consensus Scoring,” J Chem Inf Model, vol. 59, no. 9, pp. 3655–3666, Sep. 2019, doi: 10.1021/acs.jcim.9b00343.
- I. Schellhammer and M. Rarey, “FlexX‐Scan: Fast, structure‐based virtual screening,” Proteins: Structure, Function, and Bioinformatics, vol. 57, no. 3, pp. 504–517, Nov. 2004, doi: 10.1002/prot.20217.
- D. E. V Pires et al., “EasyVS: a user-friendly web-based tool for molecule library selection and structure-based virtual screening,” Bioinformatics, vol. 36, no. 14, pp. 4200–4202, Jul. 2020, doi: 10.1093/bioinformatics/btaa480.
- Maciej Serda et al., “Synteza i aktywność biologiczna nowych analogów tiosemikarbazonowych chelatorów żelaza,” Uniwersytet śląski, vol. 7, no. 1, pp. 343–354, 2013, doi: 10.2/JQUERY.MIN.JS.
- F. Gentile et al., “Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery,” ACS Cent Sci, vol. 6, no. 6, pp. 939–949, Jun. 2020, doi: 10.1021/acscentsci.0c00229.
- G. Zhou et al., “An artificial intelligence accelerated virtual screening platform for drug discovery,” Nat Commun, vol. 15, no. 1, p. 7761, Sep. 2024, doi: 10.1038/s41467-024-52061-7.
- N. Kumar and V. Acharya, “Machine intelligence-driven framework for optimized hit selection in virtual screening,” J Cheminform, vol. 14, no. 1, p. 48, Dec. 2022, doi: 10.1186/s13321-022-00630-7.
- A. Dix, S. Vlaic, R. Guthke, and J. Linde, “Use of systems biology to decipher host–pathogen interaction networks and predict biomarkers,” Clinical Microbiology and Infection, vol. 22, no. 7, pp. 600–606, Jul. 2016, doi: 10.1016/J.CMI.2016.04.014.
- T. J. Rintala, A. Ghosh, and V. Fortino, “Network approaches for modeling the effect of drugs and diseases,” Brief Bioinform, vol. 23, no. 4, Jul. 2022, doi: 10.1093/BIB/BBAC229.
- B. Ray, E. Ghedin, and R. Chunara, “Network inference from multimodal data: A review of approaches from infectious disease transmission,” J Biomed Inform, vol. 64, pp. 44–54, Dec. 2016, doi: 10.1016/J.JBI.2016.09.004.
- E. K. Silverman et al., “Molecular Networks in Network Medicine: Development and Applications,” Wiley Interdiscip Rev Syst Biol Med, vol. 12, no. 6, p. e1489, Nov. 2020, doi: 10.1002/WSBM.1489.
- Y. X. R. Wang, L. Li, J. J. Li, and H. Huang, “Network Modeling in Biology: Statistical Methods for Gene and Brain Networks,” Stat Sci, vol. 36, no. 1, p. 89, Feb. 2021, doi: 10.1214/20-STS792.
- A. A. Margolin et al., “ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context,” BMC Bioinformatics, vol. 7, no. SUPPL.1, pp. 1–15, Mar. 2006, doi: 10.1186/1471-2105-7-S1-S7/FIGURES/6.
- T. E. Chan, M. P. H. Stumpf, and A. C. Babtie, “Gene Regulatory Network Inference from Single-Cell Data Using Multivariate Information Measures,” Cell Syst, vol. 5, no. 3, p. 251, Sep. 2017, doi: 10.1016/J.CELS.2017.08.014.
- D. Beslic, M. Kucklick, S. Engelmann, S. Fuchs, B. Y. Renard, and N. Körber, “End-to-end simulation of nanopore sequencing signals with feed-forward transformers,” Bioinformatics, vol. 41, no. 1, Dec. 2024, doi: 10.1093/bioinformatics/btae744.
- K. Shafin et al., “Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads,” Nature Methods 2021 18:11, vol. 18, no. 11, pp. 1322–1332, Nov. 2021, doi: 10.1038/s41592-021-01299-w.
- G. A. Van der Auwera et al., “From FastQ Data to High‐Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline,” Curr Protoc Bioinformatics, vol. 43, no. 1, Oct. 2013, doi: 10.1002/0471250953.bi1110s43.
- S. Vadapalli, H. Abdelhalim, S. Zeeshan, and Z. Ahmed, “Artificial intelligence and machine learning approaches using gene expression and variant data for personalized medicine,” Brief Bioinform, vol. 23, no. 5, Sep. 2022, doi: 10.1093/bib/bbac191.
- M. Gokhale, S. K. Mohanty, and A. Ojha, “A stacked autoencoder based gene selection and cancer classification framework,” Biomed Signal Process Control, vol. 78, p. 103999, Sep. 2022, doi: 10.1016/j.bspc.2022.103999.
- T.-H. Zhang et al., “Transformer for Gene Expression Modeling (T-GEM): An Interpretable Deep Learning Model for Gene Expression-Based Phenotype Predictions,” Cancers (Basel), vol. 14, no. 19, p. 4763, Sep. 2022, doi: 10.3390/cancers14194763.
- J. Jumper et al., “Highly accurate protein structure prediction with AlphaFold,” Nature, vol. 596, no. 7873, pp. 583–589, Aug. 2021, doi: 10.1038/s41586-021-03819-2.
- M. Baek et al., “Accurate prediction of protein structures and interactions using a three-track neural network,” Science (1979), vol. 373, no. 6557, pp. 871–876, Aug. 2021, doi: 10.1126/science.abj8754.
- R. Wu et al., “High-resolution de novo structure prediction from primary sequence,” Jul. 22, 2022. doi: 10.1101/2022.07.21.500999.
- G. Xiong, N. J. Leroy, S. Bekiranov, N. C. Sheffield, and A. Zhang, “DeepGSEA: explainable deep gene set enrichment analysis for single-cell transcriptomic data,” Bioinformatics, vol. 40, no. 7, Jul. 2024, doi: 10.1093/BIOINFORMATICS/BTAE434.
- J. H. Oh, W. Choi, E. Ko, M. Kang, A. Tannenbaum, and J. O. Deasy, “PathCNN: interpretable convolutional neural networks for survival prediction and pathway analysis applied to glioblastoma,” Bioinformatics, vol. 37, no. Supplement_1, pp. i443–i450, Jul. 2021, doi: 10.1093/BIOINFORMATICS/BTAB285.
- M. R. Antoniewicz, “Dynamic metabolic flux analysis—tools for probing transient states of metabolic networks,” Curr Opin Biotechnol, vol. 24, no. 6, pp. 973–978, Dec. 2013, doi: 10.1016/j.copbio.2013.03.018.
- J. Reimand et al., “Pathway enrichment analysis and visualization of omics data using g: Profiler, GSEA, Cytoscape and EnrichmentMap,” Nat Protoc, vol. 14, no. 2, p. 482, Feb. 2019, doi: 10.1038/S41596-018-0103-9.
- D. S. Kolobkov, D. A. Sviridova, S. K. Abilev, A. N. Kuzovlev, and L. E. Salnikova, “Genes and Diseases: Insights from Transcriptomics Studies,” Genes (Basel), vol. 13, no. 7, p. 1168, Jul. 2022, doi: 10.3390/GENES13071168/S1.
- A. L. Mattei, N. Bailly, and A. Meissner, “DNA methylation: a historical perspective,” Trends in Genetics, vol. 38, no. 7, pp. 676–707, Jul. 2022, doi: 10.1016/J.TIG.2022.03.010.
- K. Galbraith and M. Snuderl, “DNA methylation as a diagnostic tool,” Acta Neuropathol Commun, vol. 10, no. 1, Dec. 2022, doi: 10.1186/S40478-022-01371-2.
- K. Sahoo and V. Sundararajan, “Methods in DNA methylation array dataset analysis: A review,” Comput Struct Biotechnol J, vol. 23, pp. 2304–2325, Dec. 2024, doi: 10.1016/J.CSBJ.2024.05.015.
- B. Liang, H. Gong, L. Lu, and J. Xu, “Risk stratification and pathway analysis based on graph neural network and interpretable algorithm,” BMC Bioinformatics, vol. 23, no. 1, pp. 1–13, Dec. 2022, doi: 10.1186/S12859-022-04950-1/FIGURES/4.
- B. Yuan, D. Yang, B. E. G. Rothberg, H. Chang, and T. Xu, “Unsupervised and supervised learning with neural network for human transcriptome analysis and cancer diagnosis,” Scientific Reports 2020 10:1, vol. 10, no. 1, pp. 1–11, Nov. 2020, doi: 10.1038/s41598-020-75715-0.
- G. Galindez, S. Sadegh, J. Baumbach, T. Kacprowski, and M. List, “Network-based approaches for modeling disease regulation and progression,” Comput Struct Biotechnol J, vol. 21, pp. 780–795, Jan. 2023, doi: 10.1016/J.CSBJ.2022.12.022.
- S. Aghakhani, S. E. Silva-Saffar, S. Soliman, and A. Niarakis, “Hybrid computational modeling highlights reverse warburg effect in breast cancer-associated fibroblasts,” Comput Struct Biotechnol J, vol. 21, pp. 4196–4206, Jan. 2023, doi: 10.1016/j.csbj.2023.08.015.
- M. R. Nikmaneshi, R. K. Jain, and L. L. Munn, “Computational simulations of tumor growth and treatment response: Benefits of high-frequency, low-dose drug regimens and concurrent vascular normalization,” PLoS Comput Biol, vol. 19, no. 6 June, Jun. 2023, doi: 10.1371/journal.pcbi.1011131.
- T. Rachel, E. Brombacher, S. W€ Ohrle, O. Groß, and C. Kreutz, “Dynamic modelling of signalling pathways when ordinary differential equations are not feasible,” Bioinformatics, vol. 40, no. 12, Nov. 2024, doi: 10.1093/BIOINFORMATICS/BTAE683.
- Z. Wang, L. Zhang, J. Sagotsky, and T. S. Deisboeck, “Simulating non-small cell lung cancer with a multiscale agent-based model,” Theor Biol Med Model, vol. 4, 2007, doi: 10.1186/1742-4682-4-50.
- M. Hutson, “Artificial intelligence faces reproducibility crisis,” Science (1979), vol. 359, no. 6377, pp. 725–726, Feb. 2018, doi: 10.1126/science.359.6377.725.
- E. A. Huerta et al., “FAIR for AI: An interdisciplinary and international community building perspective,” Sci Data, vol. 10, no. 1, p. 487, Jul. 2023, doi: 10.1038/s41597-023-02298-6.
- N. Sourlos et al., “Recommendations for the creation of benchmark datasets for reproducible artificial intelligence in radiology,” Insights Imaging, vol. 15, no. 1, p. 248, Oct. 2024, doi: 10.1186/s13244-024-01833-2.
- G. Yang, Q. Ye, and J. Xia, “Unbox the black-box for the medical explainable AI via multi-modal and multi-centre data fusion: A mini-review, two showcases and beyond,” Information Fusion, vol. 77, pp. 29–52, Jan. 2022, doi: 10.1016/j.inffus.2021.07.016.
- N. Khan, M. Nauman, A. S. Almadhor, N. Akhtar, A. Alghuried, and A. Alhudhaif, “Guaranteeing Correctness in Black-Box Machine Learning: A Fusion of Explainable AI and Formal Methods for Healthcare Decision-Making,” IEEE Access, vol. 12, pp. 90299–90316, 2024, doi: 10.1109/ACCESS.2024.3420415.
- M. I. Hossain, G. Zamzmi, P. R. Mouton, M. S. Salekin, Y. Sun, and D. Goldgof, “Explainable AI for Medical Data: Current Methods, Limitations, and Future Directions,” ACM Comput Surv, vol. 57, no. 6, pp. 1–46, Jun. 2025, doi: 10.1145/3637487.
- L. Longo et al., “Explainable Artificial Intelligence (XAI) 2.0: A manifesto of open challenges and interdisciplinary research directions,” Information Fusion, vol. 106, p. 102301, Jun. 2024, doi: 10.1016/j.inffus.2024.102301.
- H. Taherdoost, “Deep Learning and Neural Networks: Decision-Making Implications,” Symmetry (Basel), vol. 15, no. 9, p. 1723, Sep. 2023, doi: 10.3390/sym15091723.
- K. Vandikas, F. Moradi, H. Larsson, and A. Johnsson, “Transfer Learning and Domain Adaptation in Telecommunications,” 2025. doi: 10.5772/intechopen.114932.
- W. M. Kouw and M. Loog, “An introduction to domain adaptation and transfer learning,” Dec. 2018.
- U. Kamath, J. Liu, and J. Whitaker, “Transfer Learning: Domain Adaptation,” in Deep Learning for NLP and Speech Recognition, Cham: Springer International Publishing, 2019, pp. 495–535. doi: 10.1007/978-3-030-14596-5_11.
- E. Laparra, A. Mascio, S. Velupillai, and T. Miller, “A Review of Recent Work in Transfer Learning and Domain Adaptation for Natural Language Processing of Electronic Health Records,” Yearb Med Inform, vol. 30, no. 01, pp. 239–244, Aug. 2021, doi: 10.1055/s-0041-1726522.
- L. Zhang and X. Gao, “Transfer Adaptation Learning: A Decade Survey,” IEEE Trans Neural Netw Learn Syst, vol. 35, no. 1, pp. 23–44, Jan. 2024, doi: 10.1109/TNNLS.2022.3183326.
- T. S. Brisimi, R. Chen, T. Mela, A. Olshevsky, I. Ch. Paschalidis, and W. Shi, “Federated learning of predictive models from federated Electronic Health Records,” Int J Med Inform, vol. 112, pp. 59–67, Apr. 2018, doi: 10.1016/j.ijmedinf.2018.01.007.
- N. Rieke et al., “The future of digital health with federated learning,” NPJ Digit Med, vol. 3, no. 1, p. 119, Sep. 2020, doi: 10.1038/s41746-020-00323-1.
- B. Yurdem, M. Kuzlu, M. K. Gullu, F. O. Catak, and M. Tabassum, “Federated learning: Overview, strategies, applications, tools and future directions,” Heliyon, vol. 10, no. 19, p. e38137, Oct. 2024, doi: 10.1016/j.heliyon.2024.e38137.
- K. Daly, H. Eichner, P. Kairouz, H. B. McMahan, D. Ramage, and Z. Xu, “Federated Learning in Practice: Reflections and Projections,” Oct. 2024.
- C. Ciliberto et al., “Quantum machine learning: a classical perspective,” Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences, vol. 474, no. 2209, p. 20170551, Jan. 2018, doi: 10.1098/rspa.2017.0551.
- A. Pyrkov et al., “Quantum computing for near-term applications in generative chemistry and drug discovery,” Drug Discov Today, vol. 28, no. 8, p. 103675, Aug. 2023, doi: 10.1016/j.drudis.2023.103675.
- A. Haleem, M. Javaid, R. Pratap Singh, and R. Suman, “Exploring the revolution in healthcare systems through the applications of digital twin technology,” Biomedical Technology, vol. 4, pp. 28–38, Dec. 2023, doi: 10.1016/j.bmt.2023.02.001.
- K. Bruynseels, F. Santoni de Sio, and J. van den Hoven, “Digital Twins in Health Care: Ethical Implications of an Emerging Engineering Paradigm,” Front Genet, vol. 9, Feb. 2018, doi: 10.3389/fgene.2018.00031.
- C. Meijer, H.-W. Uh, and S. el Bouhaddani, “Digital Twins in Healthcare: Methodological Challenges and Opportunities,” J Pers Med, vol. 13, no. 10, p. 1522, Oct. 2023, doi: 10.3390/jpm13101522.
- D. C. Wynn and A. M. Maier, “Feedback systems in the design and development process,” Res Eng Des, vol. 33, no. 3, pp. 273–306, Jul. 2022, doi: 10.1007/s00163-022-00386-z.
- D. Dixon et al., “Unveiling the Influence of AI Predictive Analytics on Patient Outcomes: A Comprehensive Narrative Review,” Cureus, May 2024, doi: 10.7759/cureus.59954.
- J. Biamonte, P. Wittek, N. Pancotti, P. Rebentrost, N. Wiebe, and S. Lloyd, “Quantum machine learning,” Nature, vol. 549, no. 7671, pp. 195–202, Sep. 2017, doi: 10.1038/nature23474.
- M. Badawy, N. Ramadan, and H. A. Hefny, “Healthcare predictive analytics using machine learning and deep learning techniques: a survey,” Journal of Electrical Systems and Information Technology, vol. 10, no. 1, p. 40, Aug. 2023, doi: 10.1186/s43067-023-00108-y.

